Original Article
, Volume: 14( 1)Cloning and Expression Study of Leucoanthocyanidin Reductase Gene (CaLAR) from Camellia assamica
- *Correspondence:
- Afruza Zaman Department of Biotechnology, Tocklai Tea Research Institute, Tea Research Association, Jorhat-785008, Assam, India
Tel: +0376 236 0972; E-mail: afruzaz@gmail.com
Received Date: January 02, 2018 Accepted Date: January 29, 2018 Published Date: January 31, 2018
Citation: Zaman A, Gogoi M, Kalita MC, et al. Cloning and Expression Study of Leucoanthocyanidin Reductase Gene (Calar) from Camellia assamica. Biotechnol Ind J. 2018;14(1):158
Abstract
Leucoanthocyanidin reductase (LAR) is one of the downstream enzymes of the flavonoid pathway in plants. This gene is known to have a positive correlation with catechin accumulation, a type of flavonoid compound. The present work aimed at isolating the LAR gene from a Camellia assamica cultivar and studying its expression pattern in the shoot tips and roots through quantitative real time-polymerase chain reaction. qRT-PCR analysis showed a 0.6-fold increase in expression of LAR in roots than the shoot tips. A full-length cDNA of this gene having an ORF of 1029 bp was cloned into a heterologous host (Escherichia coli) followed by protein expression study. The expressed protein showed a molecular weight of ~ 50 kD. Further study is required to elucidate the role of LAR gene in accumulation of catechins by comparing the gene expression with catechin concentration.
Keywords
Camellia assamica; Cloning; Escherichia coli; Leucoanthocyanidin reductase; Tea
Introduction
Flavonoids are polyphenolic compounds produced as secondary metabolites in plants. They have gained importance in the recent past due to their pronounced health-benefitting properties as antioxidant, antibacterial, antiviral, anti-obesity and anti-carcinogenic agent [1,2]. Tea is one of the most important cash crops where the flavonoids are responsible for the briskness and astringency of the brew. Tea contains 750 times more flavonoid as compared to coffee, with catechins being the highest available flavonoid [3,4]. Flavonoid biosynthesis pathway of tea involves the interaction of various enzymes leading to the production of catechins, epicatechins, gallocatechins and epigallocatechins [5].
LAR, one of the key enzymes involved in the biosynthesis of proanthocyanidins, catalyses the production of the monomeric subunits, catechins [6]. It is one of the downstream genes of the flavonoid biosynthesis pathway and might have a direct influence on the accumulation of catechins. Reports showed a positive correlation between the expression of LAR gene and accumulation of total catechins in tea (Camellia sinensis) [5]. LAR gene was first isolated from Desmodium uncinatum by Tanner et al. [7]. Later, this gene was also isolated from several plant species and characterized [6,8]. Tanner et al. showed that the LAR protein was able to catalyse the conversion of leucocyanidin, leucodelphinidin and leucopelargonidin to their corresponding trans-flavan-3-ol [7]. LAR gene has been shown to possess different expression patterns in the skin and seeds of grapevine (Vitis vinifera) [9]. This might probably indicate a difference in the accumulation pattern of catechins in different organs of the same plant.
Considering the positive correlation between LAR gene expression and catechin accumulation, a study was carried out to determine the difference in expression pattern of LAR gene in different organs (shoot tips and roots) of Assam tea (Camellia assamica). A full-length cDNA of this gene was then cloned and expressed in a bacterial expression system to study the LAR protein.
Materials and Methods
Plant material
Shoot tips (two leaves and buds) and roots were collected from the S.3A/3 tea plant maintained in the germplasm collection centre of Tocklai Tea Research Institute, Jorhat, Assam. S.3A/3 is a representative of Assam tea (Camellia assamica). The collected samples were immediately stored at -80°C to prevent any loss in sample quality due to enzymatic activities.
RNA extraction and cDNA synthesis
Total RNA was extracted from 100 mg of each sample according to the protocol of Zaman et al. [10]. The integrity and quality of the extracted RNA was checked on a 1% agarose gel. The concentration and purity of the RNA was recorded in Biophotometer (Eppendorf).
1000 ng of RNA from each sample was used for the preparation of first strand cDNA using the Verso cDNA synthesis kit (Thermo Fisher Scientific Inc.). This cDNA was later amplified using specific primers for gene expression studies and cloning purpose.
Gene expression study in different tissues of the tea plant
LAR gene sequence of Camellia sinensis present in NCBI database (www.ncbi.nlm.nih.gov) was downloaded for primer designing. Gene specific primers were designed using the Primer 3 (v.0.4.0) software to generate products in the 150 bp to 200 bp range (Supplementary Table 1). Expression of the LAR gene in shoot and root samples of tea plant was estimated through quantitative RT-PCR. cDNA prepared from both shoot tips and roots were used as template to study the gene expression pattern. Quantitative RT-PCR was carried out in a Light Cycler 480 system (Roche). PCR was performed in 10 μl reaction volume containing 1X SYBR Green master mix (Thermo Fisher Scientific Inc.), 0.5 mM of each primer, 0.5 μl cDNA template and nuclease-free water to adjust the reaction volume. All the reactions were performed in triplicates.
| Serial No. | Name | Sequence |
|---|---|---|
| 1. | CaLAR_F | 5’GTTTAACTTTAAGAAGGAGATATACATATGAGGAGGTACAGCT3’ |
| 2. | CaLAR_R | 5’CTTTGTTAGCAGCCGGATCTCTCGAGTAGTGTTACACA3’ |
| 3. | CaLAR_RT_F | 5’TTCGTACGCTGAACAAATCG3’ |
| 4. | CaLAR_RT_R | 5’TATTCACTGCTGCTGCTGCT3’ |
| 5. | VEC_F | 5’CTCGAGAGATCCGGCTGCTAACAAAG3’ |
| 6. | VEC_R | 5’CATATGTATATCTCCTTCTTAAAGTTAAAC3’ |
Supplementary Table 1. Sequences of the different primers used in the study.
Analysis of gene expression was done using the ddCt method. 18S rRNA gene was considered as the reference gene against which the Ct values obtained for the LAR gene were normalized. Fold change in gene expression was calculated relative to the sho ot sample.
Full length gene amplification and sequencing
The primers for full length gene amplification were designed to contain an adaptor sequence which is the reverse complement of a homology region of the vector. This was followed by a ribosome binding site (RBS) and gene specific sequence for ligating into the vector of interest. (Figure 1) (The primer sequences have been provided in the Supplementary Table 1).

Figure 1: Diagrammatic representation of the SLIC method.
The cDNA prepared from the shoot tips was used for amplification using the full-length gene specific primer with an expected band size of 1098 bp. PCR reaction mix was prepared in 10 μl volume consisting of 0.5 μl cDNA template, 1X Taq buffer B, 1.25 mM MgCl2, 0.5 mM each of forward and reverse primers and 0.03 U Taq DNA polymerase. The reaction was carried out in a Veriti 96-well thermocycler (Applied Biosystems) with the following profile (Table 1).
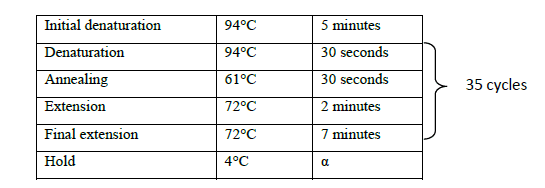
Table 1. PCR profile for full length gene amplification.
After amplification, the amplified product was electrophoresed on a 1% agarose gel at 100 V for 30 minutes. The desired amplicon was gel purified using the Hipura Quick gel purification kit (Himedia). Sequencing of the purified amplicon was done through capillary sequencing in an ABI 3130xl Genetic Analyzer (Applied Biosystems) to confirm the identity of the isolated gene.
Amplification of vector and cloning
The pACE1 vector purchased from ATG Biosynthetics, Germany was used for both cloning and expression of the LAR gene. pACE1 vector was amplified with vector specific primers (SUPPLEMETARY Table 1) according to the manufacturer’s instructions to generate the linearised form of the vector (Figure 1). Vector amplification was carried out in 100 μl reaction volume consisting of 100 ng template DNA (vector DNA), 1 mM each of forward and reverse primers, 2X HF buffer and 0.01 U Phusion DNA polymerase (NEB). The following profile was maintained during the amplification process (Table 2).
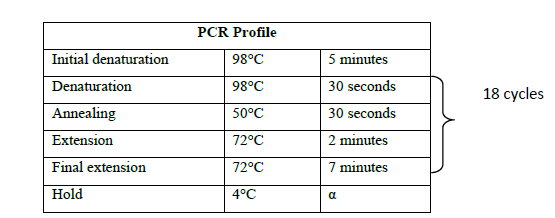
Table 2. PCR Profile for vector amplification.
The amplified linearised vector was checked on a 1% agarose gel to confirm the expected band size of 2529 bp.
The sequence and ligation independent cloning (SLIC) method as reported by Jeong et al. [11] was followed for cloning the LAR gene into the vector. This method relies on the 5’ exonuclease activity of T4 DNA polymerase to generate 3’ overhangs in the absence of dNTPs (Figure 1). The linearised vector pACE1 and the amplified LAR gene were mixed in a 1:1 molar ratio in the presence of NEBuffer 2 and T4 DNA polymerase and incubated at room temperature for 3 minutes. The reaction mix was then immediately transferred to ice and placed for 10 minutes. This mixture was used for transforming 50 μl of chemically competent DH10β cells using the heat shock method. The transformed cells were streaked on the surface of an LB agar plate containing 100 μg/ml ampicillin as a selection marker and incubated overnight at 37°C.
Protein expression study
Colonies that appeared on the selection plate were screened for the presence of the recombinant vector containing the gene of interest using colony PCR and restriction digestion. The positive clones were further used for isolation of the recombinant plasmid and then transforming the plasmid into chemically competent BL21 (DE3) pLyss cells (NEB). Protein expression was induced with 1 mM isopropyl β-D thiogalactopyranoside (IPTG) and the proteins produced were extracted using the Qproteome bacterial protein prep kit (Qiagen). The extracted protein sample was loaded on an SDS-PAGE gel to analyze the presence of the recombinant protein [12].
Results
Expression analysis of the LAR gene in shoots and roots
The total RNA extracted from the shoot tips and roots was found to be of good quality showing the presence of intact 28S and 18S ribosomal RNA bands (Figure 2). A desired A260/280 ratio of 1.8 also confirmed the quality of the extracted RNA. First strand cDNA was synthesized from 1000 ng of RNA from each sample which was later used as a template for amplification of the LAR gene.

Figure 2: Total RNA isolated from shoot tips and roots.
A part of the LAR gene was amplified in both shoots and roots of Camellia assamica plant using RT primers. A size of ~ 150 bp confirmed the exact amplification of this gene. Expression pattern of the LAR gene was then studied in both the plant parts through qRT-PCR to get an idea about the possible accumulation of catechins in the different organs. Analysis of the amplified gene in both the samples showed that the expression of the LAR gene was 0.6-fold higher in the root sample than the shoot tips. This increased expression of the LAR gene could be attributed to a higher copy number of this gene in the roots than in the shoot tips under the same environmental conditions.
Confirmation of the LAR gene sequence
Full length amplification of the LAR gene was done using gene specific primers to get a band size of 1098 bp (Figure 3). The amplified gene was sequenced and then subjected to multiple sequence alignment (MSA) using the ClustalW program with the template LAR gene sequence of C. sinensis from which the primer was designed. MSA showed more than 99% similarity with the template sequence which confirmed the identity of the LAR gene isolated from C. assamica.

Figure 3: Full length amplification of LAR gene from cDNA.
Lane 1: Amplified LAR gene of shoot tips
Lane 2: Amplified LAR gene of roots
Lane 3: 1 Kb molecular ladder (NEB).
Cloning and expression study of LAR gene
A full-length LAR gene was successfully amplified using gene specific primers and cloned into the pACE1 vector using the SLIC method. This was evident from the colony PCR results showing the amplification of LAR gene fragment in the isolated colonies (Figure 4).

Figure 4: Colony PCR of LAR gene from different colonies.
Lane 1: 500 bp molecular ladder (Genei)
Lane 2-6: LAR gene amplification from 5 different colonies
Restriction mapping was done using HindIII and BamHI restriction endonucleases to release the insert of 1098 bp and the vector backbone of 2529 bp (Figure 5).

Figure 5: Restriction digestion of the recombinant pACE1 vector releasing the insert and vector backbone.
After confirming the integration of the LAR gene into the vector, the recombinant vector was re-transformed into BL21 (DE3) pLyss expression system for the production of the recombinant protein. Recombinant protein expression was induced with 1 mM of IPTG. Total protein was extracted from the culture and loaded onto an SDS-polyacrylamide gel. The induced recombinant protein could be visualized as a dark band after staining and de-staining of the gel (Figure 6). The protein band corresponded to a size of ~ 50 kD.

Figure 6: SDS-PAGE showing the expressed recombinant LAR protein.
Lane T: LAR protein induced with 1 mM IPTG
Lane M: Protein molecular marker (Thermo Scientific).
Discussion
Tea plant serves as one of the richest source of catechins and have undergone various research related to the improvement of catechin production. The pathway for catechin production involves the communication of many enzymes of which the downstream enzyme leucoanthocyanidin reductase (LAR) plays an important part. This gene is known to be involved in the direct conversion of leucocyanidins to catechins. CaLAR gene was isolated from a C. assamica cultivar S.3A/3 and the identity was confirmed through sequencing. CaLAR had an ORF of 1029 bp which was of the same size as reported for the LAR sequence by Lei et al. [13]. The confirmation of the LAR protein was made through SDS-PAGE analysis after induction with 1 mM IPTG. A size of nearly 50 kD of this induced protein indicated the different post-translational modifications which might have added to a larger size of the protein as compared to its theoretical size of 37.5 kD. In contrast, Lei et al. [13] in an earlier study has reported a size of 60 kD for this same protein. Successful expression of the LAR gene in a heterologous host needs to be compared with the amount of catechin produced. This will enable the efficient production of catechins by manipulating the expression of the LAR gene.
LAR gene is known to positively affect total catechin accumulation. There have been many studies on the expression pattern of this gene in relation to the total catechin accumulation [6,14]. In a study conducted by Wen et al. on the effect of UV-C irradiation on flavanol accumulation, it was found that UV-C irradiation increased the synthesis and activity of LAR gene and thereby caused increased accumulation of flavanols in grape berry [15]. Liu et al. had earlier reported an Arabidopsis ldox mutant that over-expressed the cocoa LAR gene, showed elevated levels of both catechin and epicatechin [16]. In this context, we made an effort to study the expression pattern of LAR gene in the shoot tips and roots of a Camellia assamica cultivar S.3A/3. Based on the data obtained from real time – PCR, it was clear that there was a preferential expression of the LAR gene in roots than shoot tips. The over-expression of a LAR gene in roots of poplar plant (Populas trichocarpa) was reported by Yuan et al. [17] and the constitutive expression of this gene would be able to promote enhanced resistance to fungal pathogens. In tea (Camellia sinensis), the role of LAR gene in catechin accumulation in vitro was clearly analyzed by Pang et al. [18]. Later, Liu et al. also showed that the relative expression level of LAR gene in spring harvested tea plants (C.sinensis) was significantly and positively correlated with increased total catechin accumulation [19]. The tissue-specific and increased expression of the LAR gene in roots of Assam type tea plant can, as such, be an indication of the possible increased accumulation of catechins in the roots as seen from earlier reports. Having said this, further study on the concentration of catechins in shoots and roots needs to be carried out to ascertain the direct correlation of LAR gene expression with catechin accumulation.
Conclusion
A thorough understanding on the activities of the flavonoid pathway enzymes would enable us to engineer the pathway genes for improving the catechin production. LAR gene, an important downstream gene of the pathway, has previously been shown to positively effect catechin accumulation. An increased expression of the LAR gene was seen in the roots of Camellia assamica tea plant as compared to the shoot tips. This could possibly indicate a higher catechin content in the roots which needs to be further evaluated. Successful cloning of a full-length LAR gene and expression in a bacterial host would create an opportunity for the heterologous production of catechins in the future for commercial purposes.
References
- Nijveldt RJ, van Nood E, van Hoorn DE, et al. Flavonoids: A review of probable mechanisms of action and potential applications. The American Journal of Clinical Nutrition. 2001;74(4):418-25.
- Pal DK, Verma P. Flavonoids: A powerful and abundant source of antioxidants. International Journal of Pharmacy and Pharmaceutical Sciences. 2013;5(3):95-8.
- Tea and Herbal Association of Canada. Tea for Life [Online]. Available from http://www.tea.ca/iced-tea-month/tea-for-life/. [Accesed June 2017].
- Punyasiri PA, Abeysinghe IS, Kumar V, et al. Flavonoid biosynthesis in the tea plant Camellia sinensis: Properties of enzymes of the prominent epicatechin and catechin pathways. Archives of Biochemistry and Biophysics. 2004;431(1):22-30.
- Zhang LQ, Wei K, Cheng H, et al. Accumulation of catechins and expression of catechin synthetic genes in Camellia sinensis at different developmental stages. Botanical Studies. 2016;57:31-8.
- Wang L, Jiang Y, Yuan L, et al. Isolation and characterization of cDNAs encoding leucoanthocyanidin reductase and anthocyanidin reductase from Populus trichocarpa. Plos One. 2013;8(5):e64664.
- Tanner GJ, Francki KT, Abrahams S, et al. Proanthocyanidin biosynthesis in plants: Purification of legume leucoanthocyanidin reductase and molecular cloning of its cDNA. The Journal of Biological Chemistry. 2003;278(34):31647-56.
- Matsui K, Hisano T, Yasui Y, et al. Isolation and characterization of genes encoding leucoanthocyanidin reductase (FeLAR) and anthocyanidin reductase (FeANR) in buckwheat (Fagopyrum esculentum). Journal of Plant Physiology. 2016;205:41-7.
- Bogs J, Downey MO, Harvey JS, et al. Proanthocyanidin synthesis and expression of genes encoding leucoanthocyanidin reductase and anthocyanidin reductase in developing grape berries and grapevine leaves. Plant Physiology. 2005;139:652-63.
- Zaman A, Gogoi M, Borchetia S, et al. Comparative assessment of different protocols for isolation of total RNA from various organs of the tea plant (Camellia sinensis). Research Journal of Life Sciences, Bioinformatics, Pharmaceutical and Chemical Sciences. 2016;2(3):95-107.
- Jeong JY, Yim HS, Ryu JY, et al. One step sequence and ligation independent cloning as a rapid and versatile cloning method for functional genomics studies. Applied and Environmental Microbiology. 2012;78(15):5440-43.
- Sambrook J, Russell W. Molecular cloning: A laboratory manual. 3rd ed. Cold Spring Harbor Laboratory Press; New York; 2001.
- Lei MC, Yan QX, Liang C. Cloning and expression analysis of leucoanthocyanidin reductase gene of tea plant (Camellia sinensis). Journal of Tea Science. 2010;30(1):27-36.
- Wei K, Wang L, Zhou J, et al. Catechin contents in tea (Camellia sinensis) as affected by cultivar and environment and their relation to chlorophyll contents. Food Chemistry. 2011;125(1):44-8.
- Wen PF, Ji W, Gao MY, et al. Accumulation of flavanols and expression of leucoanthocyanidin reductase induced by postharvest UV-C irradiation in grape berry. Genetics and Molecular Research. 2015;14(3):7687-95.
- Liu Y, Maximova S, Payne MJ, et al. Proanthocyanidin synthesis in Theobroma cacao: Genes encoding anthocyanidin synthase, anthocyanidin reductase and leucoanthocyanidin reductase. BMC Plant Biology. 2013;13:202-20.
- Yuan L, Wang L, Han Z, et al. Molecular cloning and characterization of PtrLAR3, a gene encoding leucoanthocyanidin reductase from Populus trichocarpa and its constitutive expression enhances fungal resistance in transgenic plants. Journal of Experimental Botany. 2012;63(7):2513-24.
- Pang Y, Abeysinghe ISB, He J, et al. Functional characterization of proanthocyanidin pathway enzymes from tea and their application for metabolic engineering. Plant Physiology. 2013;161:1103-16.
- Liu M, Tian HL, Wu JH, et al. Relationship between gene expression and the accumulation of catechin during spring and autumn in tea plants (Camellia sinensis L.). Horticulture Research. 2015;2:15011-8.

