Review
, Volume: 19( 1)Overview of Biotechnological Products: Their Pharmacokinetics and Pharmacodynamics Consideration
Janhavi Mishra*
Department of Biotechnology, University of Mumbai, Mumbai, Maharashtra, India
- *Correspondence:
- Janhavi Mishra
Department of Biotechnology, University of Mumbai, Mumbai, Maharashtra, India
Tel: 9757268835
E-mail: janhavi.mishra@hkcp.edu.in
Received: September 21, 2022, Manuscript No. TSBT-22-75378; Editor assigned: September 23, 2022, PreQC No. TSBT-22-75378 (PQ); Reviewed: October 07, 2022, QC No. TSBT-22-75378 (R); Revised: January 10, 2023, Manuscript No. TSBT-22-75378; Published: January 18, 2023, DOI: 10.37532/0974-7435.23.19.001
Citation: Janhavi Mishra. Overview of Biotechnological Products: Their Pharmacokinetics and Pharmacodynamics Consideration. Biotechnol Ind J. 2023;19(1):001
Abstract
Biotechnology derived drugs (biotech drugs) include large molecules like soluble proteins, monoclonal antibodies and antibody fragments as well as smaller peptides, antisense oligonucleotides and DNA preparations for gene therapy. Over the past two decades, a growing portion of pharmaceutical R&D has been devoted to biotech drugs. For biotech drugs, choosing the best route of administration necessitates a thorough understanding of their absorption characteristics that go beyond physicochemical properties. These characteristics include expression levels, passage through biomembranes, chemical and metabolic stability at the absorption site and active uptake and exsorption processes. Therefore, it is expected that pharmacokinetic and pharmacodynamic approaches will continue to play a vital role in the successful advancement of therapeutic products derived from biotechnology in the future. The same pharmacokinetic and pharmacodynamic principles that apply to conventional small molecule pharmaceuticals medications also apply to biotechnological pharmaceuticals like peptides, proteins and oligonucleotides. Furthermore, there are recommendations for PK/PD related drug development, such as those in the recently released exposure response guidance document of the US food and drug administration and the ICH E4 guideline of the international conference on harmonization of technical requirements for registration of pharmaceuticals for human use.
Keywords
Protein; Peptides; Oligonucleotide; Monoclonal antibodies; Antisense oligonucleotides
Introduction
Pharmaceutical biotechnology
The use of living things or their components in the production of goods, as well as the enhancement of plants, animals, and microorganisms, constitutes the comprehensive scientific research area known as biotechnology. One industry that produces cutting edge goods and technology is medicine (development of new medicines and therapies). Research and development (R and D) efforts in the pharmaceutical industry have recently been concentrated primarily on biotechnologically derived drugs (biotech drugs), such as proteins, peptides, monoclonal antibodies and antibody fragments, as well as antisense oligonucleotides and DNA preparations for gene therapy. On the other side, biotechnology has highly improved the medical area. Pharmaceutical biotechnology, which applies biotechnology ideas to medication discovery, is a relatively new and fast expanding field.
Proteins and peptides
A peptide bond holds the linear chain of amino acids that make up proteins, a massive chemical complex, together. Protein is an amino acid chain that contains more than 50 amino acids. The brief polymer known as a peptide is created when less than 50 amino acids are linked together. The number, type and sequence of amino acids that make up a polypeptide chain are referred to as the primary structure of a protein. In this structure, the N and C terminals of each amino acid are always displayed on opposite sides of the polypeptide chain. The insulin molecule is the best example of the primary structure. The secondary protein structure in which long polypeptide chains are folded or stacked in various geometric configurations. Proteins have two different secondary structural arrangements: Beta pleated sheets and alpha-helical sheets. The three dimensional coiling and folding of the chain that occurs in the tertiary structure of proteins is stabilized by the interactions between amino acid sequences; this folding leaves the (R-) group on the side of the amino acids and these interactions are primarily (H-) bound interactions. The final form of a protein’s tertiary structure can be elongated, globular or any other atypical shape. Proteins have a quaternary structure that is created when two or more polypeptide chains are held together by non-covalent bonds.
Literature Review
Barriers in protein and peptide therapy
Biochemical barrier: Two major categories of biochemical barriers to the bioavailability of orally ingested proteins exist: Enzymatic and pH. The stomach and intestine possess the most active biochemical barriers to the bioavailability of orally ingested proteins because proteins are macromolecules and easily degrade in the stomach. Therefore, when proteins are administered orally, there is no activity of absorption. Gastric glands release digestive fluids, which are made up of hydrochloric acid, the protein digesting enzyme pepsin and mucus. The stomach is the body's most acidic organ because to hydrochloric acid (pH 1-2). Pepsin functions best in such very acidic environments. Pepsin is a wide endopeptidase that is abundant in the stomach and hydrolyzes the peptide bonds of aromatic residues including phenylalanine, tryptophan and tyrosine [1].
Mucus barrier: Goblet cells release mucus, a viscoelastic, hydrogel like material that lines the digestive canal. Mucins, a family of highly glycosylated glycoproteins with a predisposition to form gels due to their charged, bottlebrush like topology, make up the majority of mucus. Water, lipids, electrolytes, immunoglobulins, antimicrobial peptides, protease inhibitors and a number of other active proteins are also present in mucus. Therefore, mucus serves as a barrier to harmful bacteria while providing a nutrient rich environment for commensal bacterial colonization throughout the gastrointestinal system.
Strategies to overcome these drawbacks by: Altering route of administration
Parenteral: I.V administration of proteins and peptides avoids pre systemic degradation, achieving efficient bioavailability. The FDA approved tissue Plasminogen Activator (t-PA) analogue alteplase and the recombinant human erythropoietin epoetin-α are two examples of FDA approved IV proteins. Epoetin alfa (Epogen, Procrit), a glycoprotein produced by rDNA technology, contains 165 amino acids in an identical sequence to that of endogenous human erythropoietin. Erythropoietin also affects the release of reticulocytes from the bone marrow into the bloodstream, where these mature into erythrocytes. It is approved for anemia related to cancer chemotherapy, chronic dialysis and zidovudine (AZT) therapy.
Transdermal Drug Delivery (TDD): TDD is a minimally invasive or noninvasive technique that permits a specific dose of medication to pass through the epidermal layer of the skin by free diffusion or another manner before continuing to slowly enter the systemic blood circulation.
The following benefits of TDD:
A bigger accessible region for TDD is made possible by the vast surface area of the skin, which accounts for 1/3 of the blood circulation. Additionally, the transdermal method can prevent hepatic first pass impact and gastrointestinal degradation, increasing the drug's bioavailability. It can be given over a lengthy period of time at a specific pace (lasting several hours to several days). TDD is less traumatic, has a reduced risk of infection, costs less and has a higher success rate than subcutaneous injection.
Pharmacokinetic: Traditionally, getting peptide and protein medications to the target site of action has been the most challenge for a successful treatment. Typically, oral administration of exogenously administered peptides and proteins using conventional dosage forms results in no clinically useful absorption. Most peptides and proteins have little to no action after oral administration, which has led to the use of nonoral delivery channels such as nasal, buccal, rectal, vaginal, percutaneous, ophthalmic or pulmonary in the past in addition to parenteral administration. Drug transport via various administration routes, however, is also typically accompanied by presystemic degradation mechanisms. Numerous peptides and proteins, for instance, have much lower bioavailability following subcutaneous or intramuscular delivery than they do when taken orally [2].
Distribution: Many protein therapies have tissue compartments as their site of action. Therefore, it is crucial to comprehend the factors that affect how these molecules are distributed in the body after systemic or extravascular administration. Protein is often only distributed in the body's vascular and interstitial spaces. Whereas tissue specific distribution is influenced by both the physiological characteristics of a particular tissue and the physicochemical characteristics of the molecule. One of the primary mechanisms for the extravasation of proteins into tissues through para cellular pores of the vascular endothelium is convection. To rule out tissue build-up of potentially hazardous metabolites, whole body distribution studies are crucial for conventional small molecule medications. Protein therapies do not have this issue since the endogenous amino acid pool recycles the amino acids that are produced during catabolic breakdown. Consequently, bio distribution studies for peptides and proteins are generally carried out to determine the main elimination organs as well as to evaluate targeting to particular tissues. Because of their large molecular weight and thus limited mobility due to impeded transit across biomembranes, the volume of distribution of proteins is often modest and restricted to the volume of the extracellular space. Peptides and proteins often follow a biexponential plasma concentration time profile after intravenous administration, which is best explained by a two compartment pharmacokinetic model.
Metabolism: Phase I (functionalization) and phase II (conjugation) enzymes interact with small molecule medicines. Conversely, peptide and protein medications are they are open to untargeted proteolysis and vulnerable to serum and tissue proteases. Peptides and proteins typically have low bioavailability (less than 2%) and short half-lives due to rapid renal clearance, so they must either be administered parenterally or chemically altered to increase their stability and bioavailability or they must be administered using specialized formulations.
Excretion: The endogenous amino acid pool is used to repurpose amino acids from peptide and protein medications for the de novo manufacture of structural or functional body proteins. Peptide and protein pharmaceuticals are almost completely degraded through the same catabolic mechanisms as endogenous or dietetic proteins. Non-metabolic elimination routes including biliary and renal excretion include minimal for the majority of peptides and proteins. However, certain peptides and proteins, such as immunoglobulin A, as well as amino acids are discharged into the bile. Proteins and peptides can be excreted either generally throughout the body or only from a particular organ or tissue. Intensive peptide and protein metabolism occurs in the liver, kidneys, digestive system, blood and several other bodily components. The principal metabolic site and the predominate degradation mechanism are determined by molecular weight.
Pharmacodynamic of protein therapeutic: Since protein therapeutics is tailored treatments for a particular, well defined pharmacologic structure or mechanism, they are typically very powerful substances with steep dose effect curves. Therefore, it is particularly desirable for protein treatments to carefully characterize the concentration effect connection or the pharmacodynamics. By integrating Pharmacokinetic Pharmacodynamic modeling (PK/PD modeling), which combines pharmacodynamics and pharmacokinetics, it is possible to characterize the dose exposure response relationship of a drug and continuously describe the time course of the intensity of the effects that result from the administration of a specific dosage regimen.
Monoclonal antibody
Protein molecules known as antibodies or immunoglobin are created by the B-lymphocytes, a specific type of cell found in mammals. Monoclonal Antibodies (MAb) are generated from a single B-lymphocyte clone and are monovalent antibodies that bind to the same epitope. They were produced for the first time in mice in 1975 utilizing a hybridoma method. A foreign molecule that interacts with immune system cells and causes an immunological response can serve as an antigen. Epitopes are the molecules on the antigen to which the antibodies bind. The part of an antibody that binds to an epitope is called a paratope. MAb's extremely specific binding of just one antigen determinant is what gives them their potency. As a result, mAb medicines are targeting agents and developing novel diagnostic and therapeutic approaches. Monoclonal antibodies have the ability to selectively target and deliver poisons to cancer cells, killing them while preserving healthy cells and vital lab diagnostic detectors.
Therapeutic regulation by FDA
The FDA classifies antibodies as "biopharmaceuticals," and its Center for Biologics Evaluation and Research (CBER) and Center for Drug Evaluation and Research (CDER) are responsible for approving MAbs applications (CDER). The FDA has created a "Points to Consider" paper that provides producers with guidance on concerns to take into account while developing and testing MAbs for human use, as well as details that should be included in applications for the licensing of Investigational New Drugs (INDs) or biologics. The US FDA reports that since 2008, 48 more Monoclonal Antibodies (MAbs) have been approved, bringing the total number of MAbs in therapeutic use on the international market to 61 by the end of 2017. According to data gathered from a variety of sources, such as news announcements, the database of therapeutic antibodies, corporate pipelines, and the antibody society, the US FDA approved a total of 18 novel antibodies in 2018. The FDA authorized 12 novel MAbs in 2018, making up 20% of all approved medicines. The expectation is that 50% of them will be blockbusters by 2024, with yearly revenues of at least $1 billion.
Pharmacokinetic of monoclonal antibodies
Absorption: Most commercially available antibodies are marked for intravenous use. Administration (IV), however, a number of antibodies have been authorized for administration extravascularly. For instance, omalizumab, adalimumab, efalizumab and certolizumab pegol are permitted for delivery by Subcutaneous (SC) route. Palivizumab is delivered by Intramuscular (IM) injection and ranibizumab is injected into the eye. Having antibodies oral administration has not been effectively developed; presystemic degradation limits antibody uptake in through ineffective diffusion or convection across the gastrointestinal epithelium and through the gastrointestinal tract. With that said, intravitreal injection of ranibizumab is used to the effectiveness of MAbs after extravascular delivery depending on systemic absorption to generate a localized impact. Convective transport of antibodies through lymphatic channels and into the body is the main routes for systemic absorption. The blood and antibody diffusion via blood arteries positioned close to the injection location, in real life, the ratio of presystemic catabolism to systemic absorption determines bioavailability. Presystemic catabolism may be influenced by the pace of extracellular breakdown (for example, through proteolysis), the rate of antibody endocytosis (for example, receptor mediated, fluid phase) and the rate of intracellular oxidative stress. Interaction with the Bram bell receptor of recycling (FcRn).
Distribution: The rate of extravasation in tissue, the pace of distribution within tissue, the rate and degree of antibody binding in tissue, and the rate of removal from tissue all affect how MAb’s are distributed. Large, polar molecules like MAb are diffuse extremely slowly across vascular endothelial cells and convection is thought to be the main process causing this transfer of an antibody from blood to a tissue's interstitial fluid [3,4].
Metabolism and elimination: Filtration is one of the typical medication elimination processes (For instance, into the urine), secretion (For instance, into the bile) and biotransformation (e.g. metabolism or catabolism). Renal expulsion, it is the main mechanism for the elimination of small molecules IgG's importance to medications is comparatively minor since its huge size glomerular filtration is effective release into the bile is a crucial organ for the removal of IgA antibodies, However, this method does not significantly contribute to the removal of IgG antibodies. Most IgG removal is place through fluid phase endocytosis, subsequent intracellular degradation or receptor mediated endocytosis targeted elimination is capacity constrained (saturable) by definition as a result of the target's limited expression. The rate of absorption and eviction is a function of the dosage of antibodies through target mediated mechanisms and the target's degree of expression, as well as a characteristic of the Kinetics of intracellular catabolism and receptor internalization. But it's crucial to remember that target mediated removal doesn't always need fab attachment to a cell surface receptor [5,6].
Pharmacodynamic consideration of monoclonal antibody: The pharmacological effects that medicine has on the body are referred to as Pharmacodynamics (PD). PK for small compounds often occurs without regard to PD. Due to the Target Mediated Drug Disposition (TMDD) phenomenon; MAb PK/PD connections are special and frequently result in mAb PK dependence on PD. As was already mentioned, MAb’s can target soluble or membrane bound antigens. The binding of the target antigen and the ensuing downstream consequences, as well as effector activities like ADCC and CDC, may be what triggers PD responses. Inhibition of ligand receptor interactions by binding of MAb’s to soluble targets, down modulation of target antigen by the death of target cells or influence on cell signaling by blocking receptors are a few examples of PD responses, depending on the therapeutic mAb's mode of action.
Oligonucleotides (Antisense oligonucleotide)
Nucleic acid polymers called oligonucleotides have the potential to treat or control a variety of ailments. Although gene silencing has been the main focus of most oligonucleotide therapies, alternative approaches, such as splicing modification and gene activation are being studied, broadening the universe of potential targets beyond those typically accessible to traditional pharmacological methods. When just the main sequence of a target gene is known, it is frequently possible to rationally build highly specific lead drugs using lead candidates found through fast screening. Conventional small molecule medicines, on the other hand, need significantly more intensive and frequently repeated, screening efforts, followed by comprehensive medicinal chemistry optimization. Additionally, oligonucleotides may theoretically be created to specifically target any gene with few or at least predictable, off target effects. This enables precision and/or customized treatment techniques.
Therapeutics application: The idea of using oligonucleotides as therapeutic agents was initially put out in the late 1970’s, but its medical application was delayed due to delivery, stability and specificity problems. Numerous research projects have been carried out with the goal of chemically improving oligonucleotides. To increase oligonucleotide stability in plasma, phosphodiester bonds and sugar groups have been modified to increase their resistance to nucleases, their affinity for serum proteins and their selectivity for their target sequence. To get around delivery issues and tissue selectivity, formulations and conjugations with certain chemical groups were created.
Antisense drugs: Fomivirsen/Vitravene, an ASO medication that targets the Cytomegalovirus (CMV). RNA sequence for the treatment of CMV retinitis in immune compromised individuals, particularly those with acquired immunodeficiency syndrome, was the first ASO to get approval in 1998. (AIDS A few years later, because of breakthrough Human Immunodeficiency Virus (HIV) triple treatment, the great medical demand that existed at the time the medicine was discovered and created owing to CMV emerging in AIDS patients rapidly diminished. It was originally found at the National Institutes of Health (NIH) and after being developed and licenced by Isis Pharmaceuticals (now Ionis Pharmaceuticals), who then licenced it to Novartis, it was then discovered. Sulfur is substituted for a non-bridging oxygen atom in the phosphodiester bonds joining the nucleosides in Phosphorothioate (PS) connections seen in fomivirsen S).
Pharmacokinetic of oligonucleotides antisense
Absorption: With fomivirsen being the first authorized antisense oligonucleotide medication product, antisense oligonucleotides show enormous potential as innovative therapeutic agents intended to precisely and selectively block the synthesis of disease related products. Only Phosphorothioate oligonucleotides have a sizable amount of preclinical and human Pharmacokinetic data at this time (PONs). The average oral bioavailability ranges from 1% to 3%, which is quite low. However, ongoing research suggests that the proper release of the medicine and permeability improving excipients can boost oral bioavailability. PONs has furthermore been given by pulmonary, intradermal and subcutaneous administration methods. PONs has two compartment properties in general after intravenous injection and they are quickly removed from the plasma mostly by distribution mechanisms, with a half-life of 0.5 h to 1 h depending on the dosage. For instance, the distribution half-life of the ICAM1 inhibitor alicaforsen in humans ranges from 1.0 hours to 1.2 hours. Plasma pharmacokinetics are nonlinear and the saturation of tissue uptake is most likely to blame for the greater than proportionate rise in Area Under the Curve (AUC) with dosage [7,8].
Distribution: With the exception of the brain and testes, PONs is found in virtually all tissues and organs after intravenous injection, indicating strong transport obstacles in these tissues. Both the dosage quantity and the dose pace affect how much of the dose is absorbed by the tissues. PON seems to accumulate mostly in the liver and kidneys, with smaller amounts being seen in the spleen, bone marrow and lymph nodes. However, phosphothioate backbone structural alteration by chemicals may change organ distribution and protein binding. Although the exact mechanisms for absorption into target cells have not yet been fully understood, these processes depend on energy, temperature and time and most likely include pinocytosis and podocytes [9].
Metabolism and excretion: The nuclease mediated metabolism that removes PONs from tissues has half-lives that range from 20 hours to 120 hours, depending on the organ or tissue. While both 3 and 5 exonucleases excision may occur in tissues, the predominant metabolic route in plasma is the sequential removal of bases from the 3 end. Exonuclease metabolism is quick in plasma and tissues; after 5 minutes, 30%-40% of PON had at least one nucleotide eliminated. In general, endonuclease mediated degradation of PONs is not seen.
Pharmacodynamic consideration of oligonucleotide: Pharmacodynamics refers to the interaction of medicines, such as ASOs, with their biological targets and the processes by which the medication influences the target to cause a physiological response (PD). Determining how the ASO affects target mRNA and/or target protein levels, downstream consequences that come from target modulation and therapeutic efficacy within a specific disease indication have therefore been the primary foci of preclinical and clinical research of ASO PD. Important aspects in this context include the method of target identification and validation, the period of activity required to provide a therapeutic impact, the time before action begins and the availability of clinical biomarkers to track ASO function.
Discussion
Gene therapy
The field of therapies known as gene therapy aims to treat or vastly improve the management of conditions for which there are few or no effective treatments currently available. Advanced stage cancer or hematological illnesses make up a significant share of the gene therapy prospects. Additionally, gene therapy frequently targets uncommon or hereditary diseases. Although most gene therapy advancements are still in the research phase, businesses are spending more and more on these technologies. Several products have recently received foreign approval or are at an advanced stage of clinical development. The initial price tag for gene therapy is often extremely expensive. It is vital to have multi stakeholder discussions about how to manage the pricing and reimbursement of these items. The gene therapy agent can be injected into the body (in vivo gene therapy) or used to modify cells taken from the body, which are then re infused (ex vivo gene therapy).
Vectors used in gene therapy: Natural viruses and plasmids that have undergone modification are utilized as vectors in gene therapy. By replacing disease causing genes with the gene(s) being transferred and the sequences that govern its expression, viruses have been altered while retaining the viral envelope or coat, which facilitates transmission. Small circular DNA segments known as plasmids can be enclosed in an artificial lipid membrane or polymer to enhance transmission even though they lack a natural coat or envelope. Adeno Associated Virus (AAV), a widespread and nonpathogenic tiny virus that causes upper respiratory infections, adenoviruses and herpes viruses are examples of frequently encountered DNA viruses. Retroviral vectors generated from lentiviruses (like human HIV-1) and other RNA viruses are among them. The amount of the gene or genes it can transport, the kind and dividing status of the target cells, whether the virus will insert into the target cells genome or remain separate and the antibody status of potential patients all play a role in the choice of vector. The longest lasting expression results from insertion into the genome because the gene is kept after cell division [10].
Approved gene therapies: In USA, two ex vivo and two in vivo gene therapies have received marketing approval. All four use gene transfer technologies rather than gene editing; no gene editing technologies have been approved in the US. Talimogene laherparepvec (Imlygic, Biovec, a subsidiary of Amgen) was granted conditional approval by the FDA in October 2015 for the treatment of patients with subcutaneous or lymph node melanoma that cannot be surgically removed. It consists of recombinant herpesvirus that contains specific deletions that allow the virus to replicate and lyse tumor cells, as well as a gene carrying Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF), intended to stimulate a systemic immune response against the remaining tumor and metastases. It is administered by intratumoral injection.
Pharmacokinetic consideration of gene therapies: Only cells that have been transduced with the given gene exhibit transgene expression. The effectiveness of in vivo gene transfer is therefore significantly influenced by the tissue distribution of genes. In general, the interaction between an externally administered substance and the body, which is controlled by the physicochemical and biological characteristics of the substance as well as the anatomical and physiological characteristics of the body, determines the tissue distribution of the substance. Therefore, improving the characteristics of the drug delivery system is necessary for medication targeting or by changing the body's characteristics. The blood brain barrier's osmotic opening is an illustration of the latter. It is now well recognized that plasmid DNA is a potential nonviral vector for in vivo gene transfer since Wolff revealed that transgenic products may be generated in skeletal muscle by a straightforward intramuscular injection of naked plasmid DNA. Even for systemic delivery, quick injection of a large volume of naked plasmid DNA into the systemic circulation can result in significant quantities of transgenic output. But when plasmid DNA is administered intravenously as usual, key organs exhibit undetectable transgenic expression [11].
Uptake by target: Based on the idea of clearance, the tissue distribution of a macromolecule transported by the blood may be examined pharmacokinetically. A macromolecule is taken up by the tissue by way of uptake from the plasma and efflux by way of the tissue. When the efflux process follows first order rate kinetics and the tissue absorption rate is considered to be independent of its concentration in the plasma, the change in its quantity in a tissue over time may be characterized as follows:
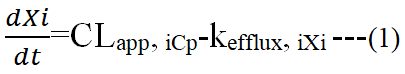
Where Xi (μg) represents an amount of the macromolecule in tissue i after administration, Cp (μg/ml) is its concentration in the plasma, CLapp, i (ml/h) expresses the apparent tissue uptake clearance from the plasma to tissue i and kefflux, i (h-1) represents theefflux rate from tissue i pharmacokinetic analysis of the macromolecule distribution process is made simpler by the assumption of negligible outflow from the tissue (kefflux, i=0). Proteins and plasmid DNA are examples of macromolecular medications that can degrade both before and after being taken up by cells.
Distribution: Capillary endothelial cells and other circulating blood cells can be directly accessed by macromolecules in the circulation. These cells have the capacity to absorb macromolecules through particular or atypical interactions. Only when macromolecules gain access to the endothelium lining can they interact with parenchymal cells in tissues. Depending on the organ, the blood capillary wall's composition varies substantially. Additionally, pathological conditions like inflammation may alter the structure. The capillary endothelium can be categorized into continuous, fenestrated and discontinuous endothelium based on the morphology and continuity of the endothelial layer and the basement membrane. The continuous endothelium, through which macromolecule movement is severely constrained, is characterized by tight connections between endothelial cells and the underlying uninterrupted basement membrane. Skeletal, cardiac and smooth muscles as well as the lungs, skin and subcutaneous tissues all have this kind of endothelium. Due to the endothelium's role as a barrier, macromolecules with a diameter of 6 nm or larger barely interact with parenchymal cells in these tissues. In the intestinal mucosa, the endocrine and exocrine glands, the glomerulus and the peritubular of the kidney, endothelial cells with fenestrae furnished with a diaphragm and an aperture 40 nm-80 nm in diameter create the fenestrated endothelium. The liver quickly takes up naked plasmid DNA. Following intravenous injection of fluorescein labeled plasmid DNA in mice, cell fractionation and confocal imaging revealed that sinusoidal cells including Kupffer cells and endothelial cells primarily take up plasmid DNA. Because plasmid DNA's significant negative charge appears to be a mediator of the uptake by these cells.
Metabolism and excretion: It is crucial to cover the negative charge of plasmid DNA in order to limit its tissue distribution because it has a very high clearance. Gal-PLL/32P plasmid DNA complex was intravenously injected and the hepatic uptake clearance was significantly higher than that of any other tissue. The pharmacokinetics of the plasmid DNA complex, however, was significantly impacted by the physicochemical characteristics of Gal-PLL utilized for the complexation. According to the clearance values, complexes with a larger Gal-PLL (13 or 29 kDa for the molecular weight of PLL) have a greater hepatic (target) clearance than those with a smaller Gal-PLL (1.8 kDa), which failed to effectively deliver plasmid DNA to hepatocytes due to complex dissociation before reaching the target.
Conclusion
Given that the biotech business is still in its infancy, there is a great need for improved methodologies. Recently, pharmaceutical corporations have made some significant strides in integrating biotechnology into their research and development procedures. This biologically based technology has tackled agriculture, sustainability and human health as well. We anticipate being able to create sufficient amounts of pure, human derived molecules in the near future and that we will be able to use these compounds to discover novel and better therapies at significantly lower costs.
References
- Bumbaca B, Li Z, Shah DK. Pharmacokinetics of protein and peptide conjugates. Drug Metab Pharmacokinet. 2019;34(1):42-54.
[Crossref] [Google Scholar] [PubMed]
- Shah DK. Pharmacokinetic and pharmacodynamic considerations for the next generation protein therapeutics. J Pharmacokinet Pharmacodyn. 2015;42(5):553-571.
[Crossref] [Google Scholar] [PubMed]
- Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576-588.
[Crossref] [Google Scholar] [PubMed]
- Ovacik M, Lin K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin Transl Sci. 2018;11(6):540-552.
[Crossref] [Google Scholar] [PubMed]
- Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies. Bio Drugs. 2010;24(1):23-39.
[Crossref] [Google Scholar] [PubMed]
- Geary RS, Norris D, Yu R, et al. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46-51.
[Crossref] [Google Scholar] [PubMed]
- Lightfoot H, Schneider A, Hall J. Pharmacokinetics and pharmacodynamics of antisense oligonucleotides. Oligonucleotide Based Drugs Ther. 2018;107-136.
- Agrawal S, Temsamani J, Galbraith W. Pharmacokinetics of antisense oligonucleotides. Clin Pharmacokinet. 1995;28(1), 7-16.
[Crossref] [Google Scholar] [PubMed]
- Nishikawa M, Takakura Y, Hashida M. Theoretical considerations involving the pharmacokinetics of plasmid DNA. Adv Drug Deliv Rev. 2005;57(5):675-688.
[Crossref] [Google Scholar] [PubMed]
- Tang L, Persky AM, Hochhaus G. Pharmacokinetic aspects of biotechnology products. J Pharm Sci. 2004;93(9):2184-2204.
[Crossref] [Google Scholar] [PubMed]
- Meibohm B, Derendorf H. Pharmacokinetics and pharmacodynamics of biotech drugs. Pharm Biotechnol. 2005;145-172.

