Original Article
, Volume: 12( 5)One-Pot Facile Synthesis of Exclusive N1-Alkyl Benzotriazoles through Intramolecular Cyclization
- *Correspondence:
- Somesh S, Department of Chemistry, HD Jain College Ara, Veer Kunwar Singh University, Ara, Bihar, India, Tel: 91-9453143260; E-Mail: someshschem@gmail.com
Received: September 07, 2016; Accepted: September 24, 2016; Published: October 06, 2016
Citation: Somesh S, Prasad V, Madhuri K, et al. One-Pot Facile Synthesis of Exclusive N1-Alkyl Benzotriazoles through Intramolecular Cyclization. Org Chem Ind J. 2016;12(5):105.
Abstract
The drawbacks associated with common synthetic approaches for N1-substituted benzotraizoles like involvement of unstable benzyne intermediate, low reaction yields, strictly anhydrous conditions and moreover the hazardous nature of low molecular weight azides prompted us to search a simple, short, and high yielding methodology to generate diverse N1-substituted benzotriazoles. Herein, a facile and high yielding protocol for diverse N1-alkyl/aryl benzotriazoles through intramolecular cyclization of different N-alkyl o-phenylenediamine/o-chloro-1,2,3-benzotriazenes under the catalysis of NaOAc.3H2O/CuI has been devised.
Keywords
Benzotriazole; Diazonium salt; Cyclization; N1-alkylation; Cyclization
Introduction
Benzotriazole containing molecules have earned the considerable attention and are in the exponential demand due to their interesting chemistry and tremendous chemotherapeutic values [1-13]. Particularly the N1-alkylated benzotriazoles are in burgeoning demand as they show exceptionally high binding affinities to a range of proteins [14] and have been recognized as potential antifungal, antibacterial, analgesic, anti-inflammatory, and antihypertensive agents [15]. Apart from the pharmacological importance, benzotriazole has been proved to be a most valuable versatile synthetic auxiliary due to several advantages over other synthetic auxiliaries and led to great deal of effort for the development of pharmacologically important benzotriazole derivatives through a simple, novel, and efficient methodology.
The common synthetic approaches for benzotriazole and their derivatives (4), generally involve the reaction of azides (2) with benzyne (3) through CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition through click chemistry ( Scheme 1), suffer due to harsh reaction condition and unstable benzyne intermediate thus, limiting the exploration of these reactions [14,16-23]. Recently developed other methods also include the hazardous azide-benzyne cyclo-addition triggered by fluoride in the presence of CsF which impedes it usefulness [14,19-20]. Similar strategy was employed to achieve the triazoles with fused aromatic skeleton via 2,3-didehydronaphthalene intermediate [21]. Synthesis of N-alkylated benzotriazoles was also achieved through reaction of alcohols with benzotriazole under Mitsunobu reaction condition [22] or benzotriazole with alkyl halides in presence of base including NaOH [23] or K2CO3 either in presence or absence of SiO2/TBAB both under the thermal as well as microwave irradiation [15,24], where mixture of N1- and N2-alkyl benzotriazoles observed. Solid-phase synthesis of 1H-benzotriazoles using Hartwig-Buchwald amination is another recent development in this promising area but unfortunately it also encountered some drawbacks [25].

Scheme 1: CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition through click chemistry.
Despite many advantages of above described methodologies for the synthesis of functionalized benzotriazoles, they experience some limitations as well, like use of hazardous chemicals, long reaction times, low reaction yields, requirement of absolute anhydrous conditions, limited availability of starting materials, harsh reaction conditions, less stability of involved benzyne intermediate, and moreover the hazardous nature of lower molecular weight azides which limit their exploration. Herein, a facile and high yielding on-pot protocol for diverse N1-alkyl-benzotriazoles through intramolecular cyclization of N-alkyl-o-phenylenediamine under the catalysis of NaOAc.3H2O has been described.
Results and Discussion
Limitations of these procedures in combination with our ongoing research on benzotriazole mediated novel synthetic methodologies [26-37] prompted us to envision a novel, simple and facile protocol for the synthesis of a variety of N1- alkyl/aryl benzotriazole derivatives under mild reaction condition without using the hazardous chemicals which impose the serious health problems. Therefore, realizing the importance of easy handling as well as green aspect of reaction, we began our synthetic strategy with readily available and environmentally benign o-chloroaniline (5), which on diazotization and in situ treatment with different aromatic amines (6) including p-anisidine, 2-napthylamine, p-toluedine, and aniline afforded the intermediates o-chloro-1,2,3-benzotriazenes in good yields. The benzotriazenes on further treatment with CuI in presence of Cs2CO3 led to the formation of various N1-aryl benzotriazoles (7) in good yields (Scheme 2) [27]. Thus, the synthesis of N1-aryl benzotriazoles was achieved in two steps. Step 1 includes the formation of intermediate o-chloro-1,2,3-benzotriazenes through diazotization and in situ coupling of amines whereas, step 2 involves the formation of desired N1-substituted benzotriazoles through N-arylation of intermediates.

Scheme 2: Benzotriazenes on further treatment with CuI in presence of Cs2CO3 led to the formation of various N1-aryl benzotriazoles.
Synthetic methodology of N-aryl benzotriazoles was standardized by studying the various parameters such as solvents (like dichloromethane, chloroform, DMF and toluene) and bases (K2CO3 and Cs2CO3) and result revealed DMF as a best suited solvent for N-arylation in combination with Cs2CO3. In order to enhance the yield further, some more bases like BABCO or DBU along with chelating ligands such as 1,10-phenanthroline or 1,8-naphthyridine were screened for the cyclization reaction of o-chloro-1,2,3-benzotriazene but could not provide the encouraging result. Considering the synthetic potential of polyvalent iodine reagents in organic synthesis [28-37], some reactions were carried out in the presence of hypervalent iodine reagent, diacetoxyiodobenzene with an aim to enhance the yield but result turned out to be unsatisfactory. All the above studies suggested that the CuI in combination with Cs2CO3 in anhydrous DMF is well suited for intramolecular N-arylation reaction. This methodology was proved to be very facile and high yielding for the easy access of N1-aryl benzotriazoles but unfortunately it failed to afford N1-alkylated benzotriazoles which are of paramount importance from pharmacological point of view. Therefore, coupling reaction of o-chloroaniline with aliphatic amines was carried out under microwave heating with aim to prepare the N1-alkylated benzotriales but reaction turned out to be unsuccessful and could not fetch the product [38,39].
The failure of above methodology to afford N1-alkyl benzotriazoles with the growing demand for alkyl benzotriazoles stimulated us to develop a simple and high yielding method for easy access to N1-alkyl benzotriazoles. The ophenylenediamine and 4-chloro-o-phenylenediamine were used as precursors for the synthesis of various N1-alkyl benzotriazoles. In first study, o-phenylenediamine was treated with different aliphatic halides (10) including benzyl chloride, 1-bromo tetradecane, butyl bromide, p-methoxy benzyl chloride, and furfuryl chloride (obtained from furfuraldehyde through known method) in anhydrous chloroform in presence of sodium hydride to afford the corresponding intermediates (11-20). These intermediates on subsequent diazotization and in situ intramolecular cyclization under the catalysis of sodium acetate trihydrate led to the formation of various N1-alkylated benzotriazoles (21-30) in very high yields (Table 1).
| R | R1 | Products (21-30) | Yield (%) |
|---|---|---|---|
| H | H | 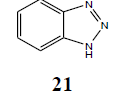 |
98 |
| H | PhCH2 | 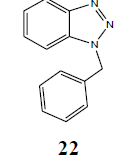 |
93 |
| H | CH3(CH2)12CH2 | 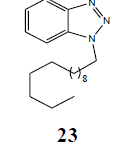 |
96 |
| H | CH3CH2CH2CH2 | 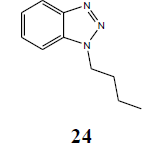 |
98 |
| H |  |
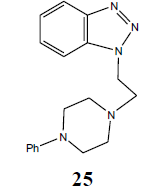 |
95 |
| H |  |
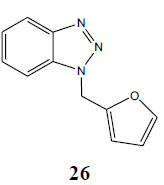 |
94 |
| H | p-MeOPhCH2 | 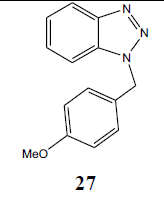 |
94 |
| Cl | PhCH2 | 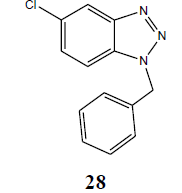 |
91 |
| Cl | PhCH2 | 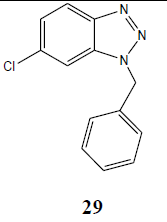 |
Obtained in trace amount |
| Cl | H | 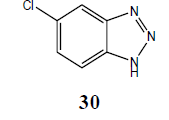 |
92 |
Table 1: Alkyl benzotriazoles (21-30) via intramolecular cyclization of N-alkyl-o-phenylenediamine.
The coupling reaction of o-phenylenediamine with various alkyl halides was considered as tricky reaction and examined intensively. Since o-phenylenediamine bears two amino groups, we presumed the coupling of alkyl halides with both amino groups and added alkyl halides in one molar ratio to diminish the any possibility of coupling with another amino group. Surprisingly, reaction furnished single product, demonstrated by TLC and later on evidenced through spectroscopic analysis (1H NMR & 13C NMR). Perhaps, steric hindrance came into the play and paved the way for the synthesis of sterically more stable product. However, the coupling reaction of 4-chloro-o-phenylenediamine with alkyl halides was investigated for its regioselectivity and the result revealed that amino group at p-position with respect to the chloro of 4-chloro-ophenylenediamine was mostly preferred for the coupling with alkyl halides than the amino group at meta position, which ultimately led to the formation of N1-alkylated benzotriazole (28) as major product after the intramolecular cyclization of coupled products. The other isomer of N1-alkylated benzotriazole (29) was also obtained but in trace quantity (Scheme 3). The electron density around the nitrogen of para amino group might have relatively been intensified due to the mesomeric or resonance effect of chloro and hence the nucleophilicity, which eventually favoured product (28).

Scheme 3: Synthesis of N1-substituted benzotriazoles via intramolecular cyclization of N-alkyl-o phenylenediamine.
The reaction of alkyl chlorides and bromides with o-phenylenediamine and 4-chloro-o-phenylelnediamine was evaluated in terms of yield and result revealed that alkyl bromides were reacted with phenylenediamines in higher yields than alkyl chlorides under the same reaction condition. This can be rationalized to higher reactivity of alkyl bromides rather than alkyl chlorides. The role of tetrahydrofuran as a solvent was found to be unfavorable for coupling reaction of alkyl halide and ophenylenediamine as it afforded the coupled products in low yield (40% to 50%) after a prolong reaction. Having seeing the poor performance of tetrahydrofuron in terms of yield as well as handling, we decided to explore some other solvent to enhance the yield further significantly in reduced reaction span and investigated anhydrous chloroform. The coupling reactions in anhydrous chloroform were proceeded very smoothly and fetched the desired products in the range of 85% to 95% yields with relatively reduced reaction times and hence emerged chloroform as a solvent of choice for N-alkylation of ophenylenediamine and 4-chloro-o-phenylenediamine.
The synthesized intermediates were subjected to diazotization and subsequent intramolecular cyclization to afford the various N1-alkylated benzotriazoles. During the diazotization of o-phenylenediamine, we observed that cyclization of diazonium was decisively influenced by the pH value of the reaction mixture. Strong basic solutions must be prevented as diazonium cations were stable only under acidic and moderately basic conditions but found to be unstable and getting transformed into diazohydroxides and finally into diazotate anions under over moderate and strong basic conditions. Under the optimized reaction condition, a series of diverse N1-substituted benzotriazoles (21-30) were obtained in very high yields using different alkyl halides such as benzyl chloride, 1-bromo tetradecane, butyl bromide, p-methoxy benzyl chloride, 1-(2-bromo-ethyl)-4-phenyl piperazine, furfuryl chloride, etc. (Scheme 3). All the developed alkylated benzotiazoles were purified by flash chromatography and characterized through spectroscopic techniques including IR, 1H NMR, and 13C NMR. Spectral data of prepared N1-Substituted benzotriazoles were found to be identical with reference compound known in literature.
In conclusion, a simple, efficient and novel method has been developed for an easy access of diverse N1-alkyl benzotriazoles through intramolecular cyclization of different N-alkyl-o-phenylenediamines under the catalysis of NaOAc.3H2O. The developed protocol offers several advantages including (a) the mild reaction condition; (b) simple workup procedure; (c) high yields of the desired products; and finally (d) the use of explosive azides is avoided. Based on our knowledge this is the first approach of its kind for an easy access to various N1-alkylated benzotriazoles through intramolecular cyclization of N-alkyl-o-phenylenediamines.
Experimental
Typical experimental procedure for the synthesis of functionalized benzotriazoles
A mixture of o-phenylenediamine (0.01 mol) and NaH (0.01 mol) in dry chloroform was allowed to stir for 15 min at 40°C followed by the slow addition of benzyl chloride (0.01 mol). After the complete addition of benzyl chloride, the whole solution was kept under constant stirring for 3 h. Then the solvent was dried under reduced pressure, added to 1 ml of ice water and stirred for 15 min at 0°C. Freshly prepared cold solutions of HCl (0.01) was added slowly to above solution with constant stirring and allowed to be stirred for another 20 min followed by the dropwise addition of freshly prepared solution of NaNO2 (0.01 mol) in 1 ml of ice cold water under constant stirring at 0°C. After complete addition of NaNO2 solution, reaction was further stirred for 30 min. Diazonium salt thus precipitated was added to a freshly prepared precooled solution of CH3COONa (0.02) over a period of 5 min to 10 min under constant stirring at 0°C and solution was left for stirring up to 1 h at same temperature. Progress of the reaction was monitored by TLC using 10% EtOAc in n-hexane. The reaction mixture was filtered through Whatman 42 filter paper and washed repeatedly with 1 ml of water. Crude mass thus obtained was subjected to flash column chromatography that furnished the pure benzotriazole product N1-benzyl benzotriazole (22) in 93% yield.
Physical data of prototype compounds
N1-Benzyl benzotriazole (22): Yield 93%; Colorless solid; MS=232 (M+Na); Mp=113-115°C; IR (KBr): νmax cm-1 1630.2, 2912.8; 1H NMR (CDCl3, 300 MHz): δ=5.85 (s, 2 H, CH2), 7.25-7.40 (m, 8 H, Ar-H), 8.05 (d, J=8.4 Hz, 1 H, Ar-H); 13C NMR (CDCl3, 75 MHz): δ=52.22, 109.67, 120.03, 123.87, 127.36, 127.52, 128.42, 128.96, 132.74, 134.70, 146.29 ppm.
N1-[2-(4-Phenyl-piperazin-1-yl-) ethyl]-benzotriazole (25): Yield 95%; Colorless solid; MS=330 (M+Na); IR (KBr): νmax cm-1 1635.9, 2937.4; 1H NMR (CDCl3, 300 MHz): δ=2.63 (m, 6 H, NCH2 and 2 x>C(CH2)2 merged together of piperazine proton), 3.21 (m, 6 H, NCH2 and 2 x>C(CH2)2 merged together of piperazine proton), 6.84-6.94 (m, 4 H, Ar-H), 7.23-7.28 (m, 5 H, Ar-H) ppm.
N1-(p-methoxy benzyl) benzotriazole (27): Yield 94%, Colorless oil. MS=262 (M+Na); IR (KBr): νmax cm-1 1628.3, 2927.6; 1H NMR (CDCl3, 300 MHz): δ=3.72 (s, 3 H, CH3), 5.73 (s, 2 H, CH2), 6.83 (d, J=8.4 Hz, 2 H, Ar-H), 7.19-7.34 (m, 5 H, Ar-H), 8.03 (d, J=8.1 Hz, 1 H, Ar-H); 13C NMR (CDCl3, 75 MHz): δ=159. 40, 146.04, 132.44, 129.18,128.33, 127.08, 126.54, 123.66, 119.63, 114.08 (Ar-C), 55.02 (OCH3), 51.57 (NCH2) ppm.
5-Chloro N1-benzotriazole (30): Yield 92%; IR Brown solid; MS=176 (M+Na); mp=153°C to 154°C; (KBr): νmax cm-1 1633.1, 2922.3; 1H NMR (CDCl3, 300 MHz): δ=7.41 (d, J=8.7 Hz, 2 H), 7.89 (m, 3 H, ), 13.04 (bs, NH); 13C NMR (CDCl3, 75 MHz): δ=114.05, 116.14, 127.15, 132.72, 140.75 ppm.
Acknowledgment
We thank CISC, BHU and RSIC, CDRI for providing spectroscopic and analytical data of synthesized compounds and University Grant Commission, New Delhi, India for Grant-in-Aid.
References
- Carlini P, Bria E, Giannarelli D, et al. New aromatase inhibitors as second-line endocrine therapy in postmenopausalpatients with metastatic breast carcinoma: A pooled analysis of the randomized trials. Cancer 2005;104(7):1335-42.
- Semple G,Skinner PJ, Cherrier MC, et al.1-Alkyl-benzotriazole-5-carboxylic acids are highly selective agonists of the human orphan G-protein-coupled receptor GPR109b.J MedChe. 2006;49(4):1227-30.
- Dipesa AJ, Donahue KM, Dombroski MA,et al. Theoretical and experimental design of atypical kinase inhibitors: Application to p38 MAP kinase. J Med Chem. 2005;48(18):5728-37.
- Wu CY, King KY, Fang JM, et al.Stable benzotriazole esters as mechanism-based inactivators of the severe acute respiratory syndrome 3CL protease.ChemBiol. 2006;13:261-8.
- Katritzky AR, Jiang J, Urogdi L, et al.N-triphenylphosphorylidene-1-(benzotriazol-1-yl)methylamine, a novel synthon equivalent to +CH2NH2: The preparation of primary amines.Tetrahedron Lett. 1989;25:3303-6.
- Wender PA, Touami SM, Alayrac C,et al. Triazole photonucleases: A new family of light activatable DNA cleaving agents.J Am Chem Soc. 1996;118:6522-3.
- Caliendo G, Greco G, Grieco P, et al.Structure-affinity relationship studies on benzotriazole derivatives binding to 5-HT receptor subtypes.Eur J Med Chem. 1996;31:207-13.
- Katritzky AR, Lan X, Jason Z, et al. Synthesis and bioassay of improved mosquito repellents predicted from chemical structure.Chem Rev. 1998;98:409-548.
- Katritzky AR, Manju K, Singh SK, et al.Benzotriazole mediated amino-,amido-, alkoxy- and alkylthio-alkylation, Tetrahedron. 2005;61:2555-81.
- Katritzky AR, Rogovoy BV. Benzotriazole: An ideal synthetic auxiliary.Chem Eur J. 2003;19:4586-93.
- Katritzky AR, Lan X. Benzotriazole-mediated arylalkylation and heteroarylalkylation.Chem Soc Rev. 1994;23:363-73.
- Katritzky AR, Henderson SA. Yang B, et al.Applications of benzotriazole methodology in heterocycle ring synthesis and substituent introduction and modification.J Heterocycl Chem. 1998;35:1123-59.
- Verma AK, Kesharwani T, Singh J, et al.Copper-catalyzed tandem synthesis of indolo- and pyrrolo[2,1-a]isoquinolines, Angew Chem Int Ed. 2009;48:1138-43.
- Chandrasekhar S, Seenaiah M, Rao CL, et al.A smooth access to benzotriazoles via azide-benzyne cycloaddition.Tetrahedron. 2008;64:11325-7.
- Nanjunda SS,Sarala BG, Priya BS, et al. Microwave-assisted synthesis of N-alkylated benzotriazole derivatives: Antimicrobial studies, Bioorg Med Chem Lett. 2006;16(4):999-1004.
- Reynolds GA. The Reaction of Organic Azides with Benzyne.JOrg Chem. 1964;29(12):3733-4.
- Karine B, Adam DM,John EM. Efficient conversion of aromatic amines into azides: A one-pot synthesis of triazole linkages.Org Lett. 2007;9:1809-11.
- Zhang F, Moses JE. Benzyne click chemistry with in situ generated aromatic azides.Org Lett. 2009;11(7):1587-90.
- Shi F, Waldo JP, Chen Y.et al. Benzyne click chemistry: Synthesis of benzotriazoles from benzynes and azides, OrgLett. 2008;10(12):2409-12.
- Campbell-Verduyn L, Elsinga PH, Mirfeizi L, et al. Copper-free ?click?: 1,3-dipolar cycloaddition of azides and arynes.Org Biomol Chem. 2008;6:3461-3.
- Kitamura T, Fukatsu N, Fujiwara Y. (Phenyl)[3-(trimethylsilyl)-2-naphthyl]-iodonium triflate as a new precursor of 2,3-didehydronaphthalene. J Org Chem. 1998;63(23):8579-81.
- Katritzky AR, Oniciu CD, Ghiviriga I.The mitsunobu reaction: An alternative synthesis of 1-(primary alkyl)benzotriazoles. Synth Commun. 1997;27:1613-21.
- Katritzky AR, Kuzmierkiewicz W, Greenhil VJ. An improved method for the N-alkylation of benzotriazole and 1,2,4-trizole. Recueil Des Travaux Chimiques Des Pays-Bas-J Royal Netherlands ChemSoc. 1991;110(9):369-73.
- Khlafi-Nezhad-K A, Zare A, Parhami A, et al. Highly regioselective N-alkylation of benzotriazole under solvent-free conditions.J Iran Chem Soc. 2007;4:271-8.
- Zimmermann V, Bras S. Hartwig-Buchwald amination on solid supports: A novel access to a diverse set of 1H-benzotriazoles. J Comb Chem. 2007;9:1114-37.
- Kale RR, Prasad V, Mohapatra PP, et al. Recent development in benzotriazole methodology for construction of pharmacologically important heterocyclic skeletons. Monatsh Chem. 2010;141(11):1159-82.
- Kale RR, Prasad V, Hussain HA, et al. Facile route for N1-aryl benzotriazoles from diazoamino arynes via CuI mediated intramolecular N-arylation. Tetrahedron Lett. 2010;51(43):5740-3.
- Kale RR, Prasad V, Tiwari VK. Facile synthesis of novel glycosyl carboxamide with sugar in furanose and pyranose form using benzotriazole methodology. Lett Org Chem. 2010;7(2):136-43.
- Tiwari VK, Singh A, Hussain HA, et al. One-pot convenient and high yielding synthesis of dithiocarbamates. Monatsh Chemie. 2008;139(1):43-8.
- Tiwari VK, Kale RR, Mishra BB, et al. A facile one-pot MW approach for 3-heteroaryl-2-thioxo-2,3-dihydroquinazolin-4(1H)-one. Arkivoc. 2008;2008(14):27-36.
- Tiwari VK, Hussain HA, Mishra BB, et al. Benzotriazole mediated one pot convenient synthesis of thioquinazolinones as potential antifungal agents. Med Chem Res. 2006;15:325-7.
- Singh A, Kale RR, Tiwari VK. Benzotriazole mediated one-pot facile synthesis of N/S-glycosyl dithiocarbamates. Trends Carbohydrate Chem. 2009;1(1):80-5.
- Tiwari VK, Singh DD, Hussain HA, et al. One-pot, simple, new, and convenient synthesis of 2-Thioxo-2,3-dihydroquinazolin-4(1H)-ones.Monatsh Chemie. 2008;139:43-8.
- Kumar D, Mishra A, Mishra BB, et al. Synthesis of glycoconjugate benzothiazoles via cleavage of benzotriazole ring. J Org Chem. 2013;78:899-909.
- Kumar D, Mishra BB, Tiwari VK. Synthesis of 2-N/S/C-substituted benzothiazoles via intramolecular cyclative cleavage of benzotriazole ring. J Org Chem. 2014;79(1):251-66.
- Kumar D, Singh AS, Tiwari VK. An unprecedented deoxygenation protocol of benzylic alcohols using bis(1-benzotriazolyl)- methanethione. RSC Adv. 2015;5:31584-93.
- Singh AS, Kumar D, Mishra N, et al. An efficient one-pot synthesis of N,N'-disubstituted ureas and carbamates from N-acylbenzotriazoles. RSC Adv. 2016;6:84512-22.
- Prasad V, Kale RR, Mishra BB, et al. Hypervalent iodine reagents in carbohydrate modification. Trends Carbohydrate Res. 2009;1(4):43-9.
- Tiwari VK, Mishra BB, Mishra KB, et al. Cu-Catalyzed Click Reaction in Carbohydrate Chemistry. Chem Rev. 2016;116(5):3086-240.