Organic Chemistry: An Indian Journal
ISSN (PRINT): 0974-7516
All submissions of the EM system will be redirected to Online Manuscript Submission System. Authors are requested to submit articles directly to Online Manuscript Submission System of respective journal.
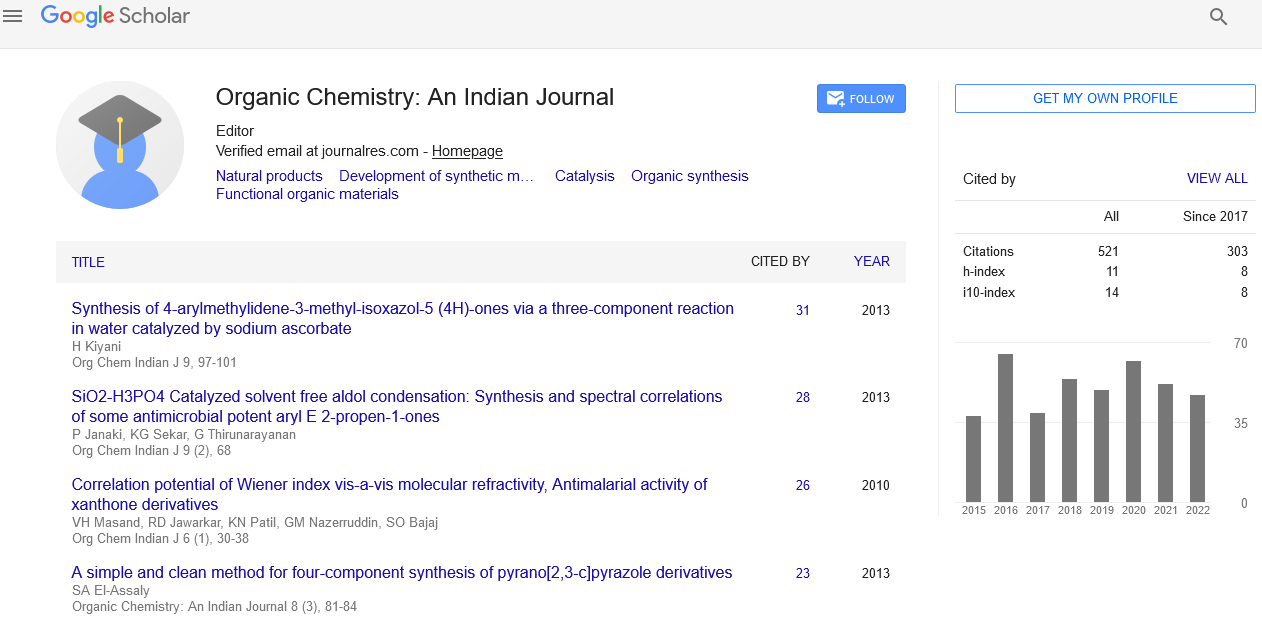
Google Scholar citation report
Citations : 565
Organic Chemistry: An Indian Journal received 565 citations as per Google Scholar report
Indexed In
- CASS
- Google Scholar
- Open J Gate
- China National Knowledge Infrastructure (CNKI)
- Cosmos IF
- Directory of Research Journal Indexing (DRJI)
- Secret Search Engine Labs
- ICMJE
View More
For Librarians
