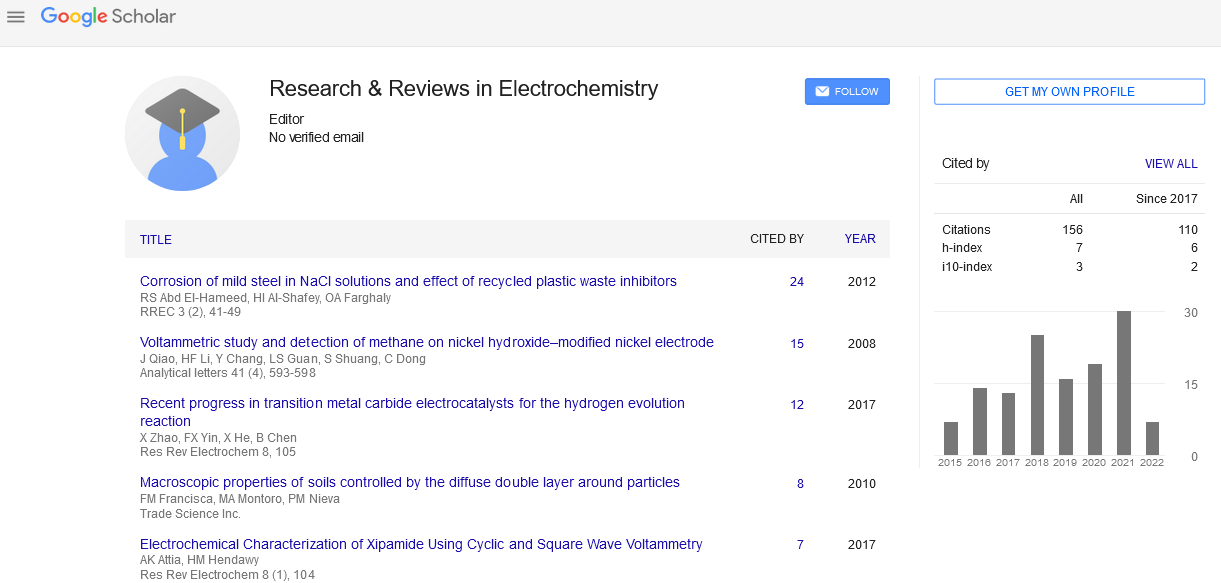
Original Article
, Volume: 9( 1)Acetaminophen Molecular Imprinted P-Phenylenediamine Glassy Carbon Electrode for Pharmaceutical Tablets Control
- *Correspondence:
- Maxime Pontié Host-Pathogen Interaction Study Group, Angers University, Brest University, Institute of Biology in Health, France, 4 rue Larrey, 49933 Angers cedex 9, France, E-Mail: maxime.pontie@univ-angers.fr
Received: August 23, 2018; Accepted: September 10, 2018; Published: September 17, 2018
Citation: Pontie M., Jaspard E., Papon N, et al. Enhanced Direct Oxidation Of Diclofenac (DCF) At A Carbon Paste Electrode (CPE) Modified With Cellulose And Its Biodegradability By Scedosporium dehoogii. Res Rev Electrochem. 2018;9(2):114.
Abstract
We report for the first time on the elaboration of a molecular imprinted polymer (MIP) based on the electropolymerization of p-phenylenediamine on a glassy carbon electrode (GCE) and dedicated to acetaminophen control in commercialized tablets. Cyclic voltammetry (CV) and square wave voltammetry (SWV) electrochemical technics are exploited to elaborate a low-cost, reproducible and very simple methodology for the determination of acetaminophen in pharmaceutical tablets. A linear evolution between the response currents at Ep=+0.52 V/SCE and acetaminophen concentration ranging from 01 mg/L to 500 mg/L is determined for MIP sensor elaborated with a limit of detection (LOD) of 30 μg/L at a signal to noise ratio (S/N) of 3. An excellent selectivity for the paracetamol vs p-nitrophenol, dopamine and dopac interferents molecules is demonstrated and recovery values obtained between 97.4% and 101.4% indicating the accuracy and repeatability of the proposed method dedicated to the quality control of commercialized tablets.
Keywords
Diclofenac detection; carbon paste electrode; biodegradation; Scedosporium; modified electrode; cellulose fibers
Introduction
Various Pharmaceutically Active Compounds (PhACs) (i.e. acetaminophen, ibuprofen, carbamazepine, diclofenac) have been frequently detected in sewage treatment plant effluents and surface waters because of the consumer use, hospital waste, and improper disposal. This finding is at the origin of a craze of the scientific community aiming at developing innovative wastewater treatment processes due to the increasing risk of contamination of the water resources dedicated to human consumption [1].
With acetaminophen, diclofenac (DCF) is one of the most used Non-Steroidal Anti-Inflammatory Drugs (NSAID) [2]. It is a highly frequently prescribed pharmaceutical compound to treat both humans and animals. Unfortunately, DCF also displays a substantial bio-accumulating potential and a chronic ecotoxicity, representing a serious risk for living organisms [3]. Therefore, DCF is now categorized as an emerging environmental micro-pollutant in the first Water Framework Directive (WFD) list of vigilance [4,5]. As recently reported in Finland [6], waste waters may contain up to 0.4 mg L-1 of PhACs.
Biodegradation of PhACs by microorganisms which have demonstrated their potential to degrade these molecules into innocuous end products such as CO2 and H2O is being increasingly considered as an environmental friendly and low-cost option. To date, treatment of these organic micropollutants in waste water is mainly based on the action of bacterial communities but, unfortunately, DCF displays a low biodegradability by bacteria. Thus a challenge remains to identify eukaryotic microorganisms capable to efficiently degrade PhACs, particularly DCF and related compounds. In this regards, fungi belonging to the genus Scedosporium progressively become attractive biodegrading agents since various Scedosporium strains were shown to be able to use aliphatic and aromatic hydrocarbons as carbon and energy sources [7-10]. Although most of the Scedosporium species are known to be opportunistic pathogens for humans, the species S. dehoogii has never been involved in deep-seated infections [11], and was extremely rarely associated to subcutaneous infections [12], making this mold a promising candidate for bioremediation purposes, as recently reported for acetaminophen biodegradation (FIG. 1)[13].

FIG 1: Quantification and bioremediation of diclofenac (DFC).
Furthermore, in the approach of health risk management, a part of the methodology suggested remains the development of new analytical tools [1]. DCF concentrations are usually determined by High Performance Liquid Chromatography, Gas Chromatography, Capillary Zone Electrophoresis, Spectrophotometry, Spectrofluorometry and Chromatography. Besides, electrochemical methods become prominent tools for monitoring PhACs compounds because they require simple operational procedures and low-cost equipment; moreover they allow quite fast analyses allowing real time measurements. In that field, chemically modified electrodes usually exploited as sensing devices are of particular interest, particularly carbon-based modified electrodes that are easy to prepare, inexpensive and generally give rise to reproducible signals [14]. Carbon materials previously used in the preparation of Carbon Paste Electrode (CPE) for DCF electroanalysis include carbon fibers, carbon powder, pyrolytic graphite, pencil graphite, Multiwall Carbon Nanotube (MWCNT), graphene or graphite powder [15-19]. Recently, ligno-cellulosic materials such as coffee husk but also pure cellulose fibers were successfully used to prepare CPE dedicated to acetaminophen determination [20-23]. Their use as modifiers aimed to increase both the real surface area and the hydrophilicity of the modified electrodes.
In the present work, we have taken advantage of a recently developed CPE modified with cellulose fibers [21] to elaborate a votametric sensor dedicated to DCF analysis as a part of the determination of DCF biodegradation by S. dehoogii.
Materials and Methods
Reagents
DCF was purchased from Sigma-Aldrich as powder in the form of sodium salt (lot#BCBV3438) and used as received. The composition of the used basal solution was as follows for 1 L of ultrapure water: 5 g (NH4)2SO4; 5 mg benonyl; 0.652 g MgSO4; 1 mg dichloran; 0.5 g chloramphenicol; 0.01 g FeSO4; 1.25 g KH2PO4. All aqueous solutions were prepared from analytical grade chemicals, using sterile deionized water obtained from an Elga Lab water ultrapure-water system (Purelab- UV-UF, Elga, France) (pH 6.5, conductivity < 1 μS cm-1 and TOC < 0.1 mol L-1).
Microorganisms and culture conditions
Five strains representative of the five prominent Scedosporium species, i.e. S. apiospermum IHEM 23577, S. dehoogii UA 110350859-01, S. boydii IHEM 4595, S. aurantiacum UA 120218507, and S. minutisporum IHEM 23833, were investigated. Strains were routinely maintained by weekly passages on Yeast Extract-Peptone-Dextrose (YPD) agar plates (1% yeast extract, 2% peptone, 2% dextrose).
The capability of Scedosporium strains to use DCF as the sole carbon source was investigated by cultivation of the fungi on Scedo-Select III agar plates containing DCF as follows. Conidia were harvested from 2-week-old cultures at 25-30°C on YPD medium plates by flooding the agar surface with 15 mL of ultrapure water. The obtained fungal suspension was then filtrated on a 40 μm pore size sterile nylon filter and conidia were pelleted from the filtrate by centrifugation at 4000 g (5 min at 4°C), resuspended in 10 mL of sterile ultrapure water and finally enumerated using an hemocytometer. Conidia were then inoculated onto Scedo-Select III agar plates [24] containing DCF as the carbon source. For this purpose, a stock solution of DCF (0.975 g DCF in 30 mL of ultrapure water with 5 ml of acetonitrile) was prepared and sterilized by filtration (0.2 μm-pore size sterile membrane). After addition of DCF to the culture medium at a final concentration of 0.9 g L-1 and inoculation with conidia (104 in 10 μL), plates were incubated during 3 weeks at 37°C for S. apiospermum, S. boydii, and S. aurantiacum, and 25°C for S. minutisporum and S. dehoogii. The radial growth was determined at day 6, 12, 16, and 21. For comparison, the same series of experiments was achieved using 4-hydroxybenzoate (4-HBz) as sole carbon source in the culture medium.
Study of DCF degradation by S. dehoogii was conducted using a cellulose-modified Carbon Paste Electrode (CPE-Ce) as follows. Conidia from 2-week-old cultures on YPD agar plates were inoculated into Yeast Extract-Peptone (YP) broth supplemented with DCF as the carbon source. After addition of DCF to YP broth at a final concentration of 0.9 g L-1, and inoculation of the conidia (106 mL-1), the culture medium was distributed into twenty 50 mL-flasks. The flasks were then incubated for two weeks at 25 ± 2°C with constant shaking (125 rpm). Fungal growth was obvious at day 3. Therefore, the residual concentration of DCF was determined from day 4 using two flasks per day. Measurements were performed using CPE-Ce by linear sweep voltammetry in the condition optimized in the first part of this work. A Control consisted in the basal medium supplemented with DCF, but not inoculated with the fungus, and incubated under the same conditions.
Apparatus
The electrochemical measurements of DCF amounts in solution were performed using an electrochemical analyzer PG580 (Uniscan Instruments, UK) connected to a personal computer. The electrochemical software used was UiEchem version 3.27, from Uniscan Instruments. A classical three-electrode cell configuration was employed, consisting of CPE-Ce or unmodified CPE, a silver/silver chloride reference electrode and a platinum wire counter electrode.
Morphological analysis of CPEs surfaces was achieved by Field Emission Gun Scanning Electron Microscopy (FEGSEM) on a JSM-6301F apparatus from JEOL (SCIAM common service in Angers University, France). A 1 cm height cylinder for each CPE tested was immobilized on a SEM sample holder using adhesive carbon tape. Images obtained were from secondary electrons of 3 keV, with magnification at 1,000 X.
Preparation of modified and unmodified CPEs, FEGSEM images of the electrodes tested and real vs geometrical surfaces determination
CPE-Ce was prepared by thoroughly hand mixing of 30 mg of silicone oil with 65 mg of graphite powder (analytical grade, ultra F, <325 mesh, from Alfa) in a mortar and 5 mg of cellulose powder. Cellulose used as CPE modifier was purchased from Fluka supplier with fibers length between 0.015 mm and 0.112 mm (lot#345768/1 595). A portion of the composite mixture was packed into the cylindrical hole of a Teflon® tube equipped with a copper wire serving as electrical contact with the rest of the circuit (FIG. 2a). The surface to be exposed to the solution was polished on a weighing paper to give a smooth aspect before use. As broadly reported in the literature [14], we have used CPEs due to their low cost and very easy regeneration procedure. We have optimized previously this aspect and demonstrated that 3 lines of 2.5 cm are necessary as illustrated in FIG. 2b and sufficient to regenerate completely the surface. The presence of roughly 10 μm diameter fibers of cellulose on the CPE-Ce compared to the CPE was revealed by FEGSEM analysis (FIG. 2c and 2d).

FIG 2: CPE design and regeneration. CPE design (a) and regeneration step of CPE with 3 lines of 3.5 cm (b) 2D-SEM images of (c) bare CPE and (d) CPE-Ce (magnification 1000 X).
For the evaluation of the real surface area of the tested electrodes, CPE-Ce and unmodified CPE were compared by cyclic voltammetry using a probe solution consisting in 5 mM [Fe(CN6)3-] in 0.1 M Phosphate Buffered Saline solution (PBS) pH 7.42. The Peak Intensity (Ip) of the probe at a given electrode can be used to determine the real surface area (A) of that electrode on the basis of Randles-Sevcik equation [25]. The geometrical surfaces of both electrodes tested, estimated to 0.071 cm2, were calculated from the area of a circle with a radius of 15 mm. By contrast the electrochemically active surface areas of CPE and CPE-Ce were evaluated from the current obtained by the probe [Fe(CN6)3-], using cyclic voltammetry. The observed peak currents measured were 126 mA and 198 mA for CPE and CPE-Ce, respectively. Using the Randles-Sevcik equation, the real geometrical areas were calculated from these peak currents to be 0.075 cm2 and 0.118 cm2, for CPE and CPE-Ce, respectively [22].
Results and Discussions
Electrochemical behavior of DCF
To determine the electroactivity domain of DCF, cyclic voltammetry (CV) was used on 1.5 mg L-1 DCF in PBS, using CPE and CPE-Ce, from -0.2 V to +0.6 V vs. Ag/AgCl. The anodic peak potential for DCF oxidation at CPEs was observed at 0.48 V as illustrated on FIG. 3a. This irreversible redox system is associated to a redox process corresponding to the direct oxidation of DCF by the equilibrium usually observed [16] and depicted in FIG. 3b.

FIG 3: Cyclic voltammetry of DCF in PBS solution at 200 mV/s of potential scan rate, DCF concentration 1.5 mg L-1, for CPE-Ce and CPE (qualitative experiment) (a). This irreversible redox system is associated to a redox process corresponding to the direct oxidation of DCF as illustrated by the equilibrium usually reported [16] (b). As a reference the CV in the absence of DCF on CPE is reported on the FIG. 2a (third curve).
But recently Aguilar-Lira et al. [17] showed that the electrochemical oxidation of DCF in aqueous media, can involve an EC mechanism with a one electron exchange reaction and a chemical reaction leading to breaking up the oxidation product through the nitrogen atom, with the formation of 2,6 dichloroaniline and 2-(2-hydroxyphenyl) acetic acid compounds. This mechanism allows to explain the presence of a reversible redox system observed between 0.00 V and 0.30 V (FIG. 3). They proposed the electrochemical reduction of 2-(2-hydroxyphenyl) acetic acid to 1-hydroxy-2-hydroxyphenyl) ethanalate and its oxidation at pH 7 with one electron transfer. These authors used this redox system to develop an original but indirect analysis of DCF based on the peak observed at 0.25 (FIG. 3). Our choice was more common because it was based on a direct analysis of DCF at the peak potential of 0.48 V.
Optimization of DCF direct oxidation
Addition of cellulose
As illustrated in FIG. 4, the oxidation current of DCF increased with the addition of cellulose fibers. We have previously determined the optimal quantity of cellulose to use [22,23]. The ratio of the peak intensities using CPE-Ce vs. unmodified CPE was estimated at 6 (result not shown). This increase in the sensitivity observed for the CPE-Ce can be attributed to increment of the geometrical surfaces due to the presence of cellulose fibers and/or the increase in the hydrophilicity of the outer layer of the CPE due to the addition of cellulose fibers. In order to investigate each hypothesis, we first determined the real CPE surface with a probe, following a classical approach in electrochemical sensor development, as recently reported [22,23]. Following this approach, we estimated with the probe Fe(CN6)3- a ratio of geometrical areas of 1.6. We observed that this ratio drastically differs from that of peak intensities obtained with DCF which has been evaluated to 6. Therefore, the difference between these two ratios corresponds to the contribution of the chemical modification of the electrode due to the addition of cellulose fibers. It is thus possible to compare the CPEs tested in terms of current density, as illustrated in FIG. 4.

FIG 4: Current density vs. potential in linear sweep voltammetry for unmodified CPE and CPE-Ce (PBS solution, pH=7.4, 2.3 mg L-1 of DCF, potential scan rate 100 mV.s-1, initial potential -0.4 V/Ag/AgCl and accumulation time 250 s)
We recently reported that the presence of cellulose fibers changes the hydrophilicity of the carbon paste [13,22,23]. Indeed by measuring a contact angle of a water droplet deposited on modified/unmodified surfaces of CPEs, we observed a decrease in the contact angle measured by the sessile drop technique, from 109° to 75° for unmodified CPE and CPE-Ce, respectively [22]. A chemical change of the surface exposed to DCF was also responsible for a 4.4 fold gain in current density when cellulose was used as modifier of the CPE. Other chemical interactions such as H-H bonds may also contribute. However, our results indicate that 27% of the current gain is due to the geometrical area increase and that the main parameter influencing this process remains above all the chemical affinity for DCF with 73% of gain. Considering the preliminary results depicted in FIG. 4, we thus selected the cellulose-modified CPE for further experimentations.
Influence of accumulation time
As shown in FIG. 5, the accumulation time significantly affected the direct oxidation peak current of DCF through changing its surface concentration on CPE-Ce. The oxidation peak current was greatly enhanced with the increase in the accumulation time within the first 300 s and then remained almost unchanged. This may be attributed to the established equilibrium of DCF concentration at the surface of CPE-Ce. This suggests that increase in real geometric area (as estimated above) seriously impacts the accumulation efficiency. On this basis, the optimal accumulation time of 250 s was chosen for the rest of the study. However, it is important to remind that the initial potential remains also a major factor which affects the voltammetry response in comparison with the other accumulation conditions, as reported by Yang et al. for [19] for DCF analysis and by Sbai et al. [26] for methyl parathion analysis using a carbon-based modified ultra-microelectrode.

FIG 5: Influence of accumulation time at CPE-Ce on the peak current of 2.3 mg L-1 DCF solution (pH 7.4) at an initial potential of - 0.4 V/AgAgCl.
Influence of initial potential
The oxidation peak current reached a maximum at - 0.4 V, as illustrated in FIG. 6. Furthermore, a dramatic decrease in peak intensity was observed for positive potentials, as reported by Yang et al. [19] on MWNTs-DHP film-coated GCE. The electric charge condition of the modified electrode would directly influence the adsorption and the oxidation of DCF involving electron and proton transfer. The optimal initial potential of - 0.4 V was determined at the maximum peak intensity.

FIG 6: Influence of initial potential on the peak current at CPE-Ce using a 2.3 mg L-1 DCF solution in 0.1 M PBS pH 7.4 at an initial potential of - 0.4 V/AgAgCl.
Application of the CPE-Ce for the study of the biodegradation of DCF by Scedosporium species
Choice of the Scedosporium species
In order to estimate the ability of Scedosporium species to metabolize DCF, a representative strain of each of the five prominent Scedosporium species including S. boydii, S. apiospermum, S. aurantiacum, S. dehoogii, and S. minutisporum was grown on Scedo-Select III medium complemented with DCF (TABLE 1).
| 4-HBz | DCF | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 6 d | 12 d | 16 d | 21 d | t1/2 (d) | 6 d | 12 d | 16 d | 21 d | t1/2 (d) | |
| S. boydii (37°C) | 0.8 | 1.1 | 1.4 | 1.5 | 8.4 | 0.2 | 0.8 | 0.9 | 1.1 | 9.8 |
| S. apiospermum (37°C) | 0.7 | 2 | 2.2 | 2.3 | 7.5 | 0.3 | 0.8 | 0.8 | 1 | 8.4 |
| S. aurantiacum (37°C) | 0.9 | 1.4 | 1.5 | 1.6 | 5.7 | 0 | 0 | 0.1 | 0.2 | 16 |
| S. dehoogii(25°C) | 1.1 | 2.2 | 2.4 | 2.9 | 10.2 | 0.5 | 1 | 1.5 | 2.4 | ND |
| S. minutisporum (25°C) | 0.8 | 1.4 | 1.7 | 1.9 | 11.2 | 0.3 | 0.8 | 1.1 | 1.5 | 32.1 |
ND : t1/2 not determinable for S. dehoogiibecause growth was still in linear phase at 21 days.
TABLE 1. Time course of radial growth and the doubling time of the culture (t1/2) of several Scedosporium species plated onto Scedo-Select III petri dishes supplemented with DCF as the sole source of carbon.
In order to compare the half-time of growth of Scedosporium species tested, data were first normalized between 0 and 1. A "Hill-like" equation (1) was used to analyse these data:
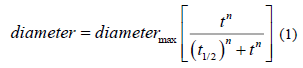
Diameter is that of the strain, measured on the Petri dish (radial growth), t is the time of the culture (in days), t1/2 is the time where 50% of the culture growth occured, n is an « apparent Hill number describing the sigmoidicity of the growth curve. Growth rates were compared to those observed onto the classical Scedo-Select III culture medium containing 4-hydroxybenzoate [24].
After addition of 4-HBz or DCF to Scedo-Select III agar mixture at a final concentration of 0.9 g L-1, and inoculation of the conidia (104 in 10 μL), plate were incubated during 21 days at 37°C for S. apiospermum, S. boydii, and S. aurantiacum, and 25°C for S. minutisporum and S. dehoogii. The radial growth of the mycelium was determined at day 6, 12, 16, and 21. As indicated in TABLE 1, S. dehoogii displayed the more rapid growth when cultivated on Scedo-Select III medium supplemented with DCF as the sole carbon source. Interestingly, this species is probably the least problematic in terms of risk to human health, as discussed recently by Blasi et al. [9]. Scedosporium dehoogii was thus selected for subsequent experiments.
Calibration plot and biodegradation study with CPE-Ce
Linear Sweep Voltammetry (LSV) was performed using CPE-Ce in basal solution in the concentration range of 0.2 mg to 2.5 mg L-1 (under the saturation occurring at 2.5 mg L-1 at 25°C). The calibration graph for DCF on the modified CPE-Ce was linear from 0.25 mg L-1 to 2.5 mg L-1. Oxidation of DCF followed the equation y = (28.3x + 21.2) where y is the peak current measured (in A) at the peak potential and x is the DCF concentration (in mg L-1) and the Limit of Detection (LOD) was determined by the method of the linear regression. The correlation coefficient (R2) was 0.999.
TABLE 2 provides the comparison of the results for the determination of DCF using different modified electrodes and various analytical parameters in the literature [15-19,27-29]. This comparative study reveals the acceptability of the present sensor over some earlier reported methods, especially in terms of LOD. This may be attributed to the immobilization of cellulose fibers at the CPE surface with enlargement of the surface area and change in the wettability of the CPE surface.
| Electrode | Methods | Linear range (µmol L−1) | Limit of detection (µmol L−1) | Reference |
|---|---|---|---|---|
| VFMCNTPE | SWV | 5–600 | 2 | 15 |
| Graphite | DPV | 2.56–9.5 | 0.76 | 17 |
| AuNPs/MWCNT/GCE | SWV | 0.03–200 | 0.02 | 18 |
| MWNTs/DHP film/GCE | LSV | 0.17–2.5 | 0.08 | 19 |
| MWCNTs/CTS-Cu/GCE | SWV | 0.3–200 | 0.021 | 27 |
| MWCNTs/Cu(OH)2/IL-GCE | LSV | 0.18–119 | 0.04 | 28 |
| rGO/CHNF/CPE | SWV | 0.025–1.55 | 0.008 | 29 |
| CPE-Ce | LSV | 0.84–8.44 | 0.02 | This work |
TABLE 2. Comparison of the analytical performance of the different modified electrodes for determination of DCF.
In order to evaluate its analytical applicability, the proposed DCF sensor was applied to the determination of a prepared solution at 1.75 mg L-1, which was by this approach estimated to 1.79 mg L-1 (recovery of 103%). The fungal inoculum was then mixed with 2 mg L-1 of DCF into 20 flasks which were stirred continuously during 12 days. From day 2, the residual concentrations of DCF into the culture medium were determined by electrochemistry analysis using the optimized CV parameters at CPE-Ce sensor. FIG. 7 shows the time course evolution of DCF concentration.

FIG 7: Evolution of ln (DCF/DCFt=0) in mg L-1 as a function of time.
DCF concentration decreased proportionally with time (FIG. 7), along with the increase in the fungal mass. This proved the degradation of DCF by the fungus and its use as a carbon source. Furthermore the linear relationship obtained between ln (DCF/DCFt=0) vs. time indicated that the process of degradation is governed by a pseudo one order kinetic (see equation (2):
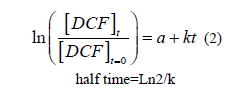
The kinetic constant k and half-life time of DCF were respectively of 0.012 day-1 and 57.8 days for an initial concentration of 1.65 mg L-1 and a temperature of 25°C.
The biodegradation pathway(s) of DCF in Scedosporium species remain fully unknown. However, previous works shed light on various biotransformation routes that could occur in microorganisms (FIG. 8). This implies in particular the intervention of different enzymes from the monooxygenase family to form mono and dihydroxy-DCF metabolites along with a pbenzoquinone imine derivative of 5-hydroxy-DCF [30]. Recently, in the course of full degradation of DCF by a forest soil microbial consortium, Facey et al. [31] identified a carboxylated diclofenac intermediate by LC-MS/MS-TOF. This might be a key intermediate to enable a complete biodegradation of diclofenac via 2,6-dichloroaniline and carboxylated 2- hydroxyphenylacetic acid through 2-chloromaleylacetate and 4-maleylacetoacetate, respectively (FIG. 8). Of course, further experiments are needed to decipher the full catabolic pathway of DCF in Scedosporium species.

FIG 8: Putative DCF degradation intermediates in Scedosporium spp. previously characterized catabolites in various microorganisms are drawn in velvet and unknown intermediate are in grey. In nature, the degradation of DCF by a group of white rot fungi (Phanerohaete spp.) has mainly been demonstrated. Two mono-hydroxylated metabolites, one di-hydroxylated DCF catabolites and one p-benzoquinone imine derivative of 5-hydroxy-DCF were identified as degradation intermediates in fungal cultures. More recently, Facey et al. [31] isolated a carboxylated diclofenac intermediate. This might be a key intermediate to enable a complete biodegradation of diclofenac via 2, 6-dichloroaniline and carboxylated 2-hydroxyphenylacetic acid by a microbial consortium. Putative degradation pathways of both 2, 6-dichloroaniline and carboxylated 2-hydroxyphenylacetic acid are proposed. R corresponds to a carboxyl group.
Conclusion
This study proposed a novel CPE modified with cellulose fibers and optimized for DCF electroanalysis. It was used for the first time to determine the kinetic parameters of DCF biodegradation by Scedosporium species. This study has shown the potential of using cellulose powder to increase the geometrical area and in the same time the hydrophilicity of the unmodified CPE. The optimized accumulation time was 250 s and initial potential - 0.4 V/Ag/AgCl/Cl-. Among the prominent Scedosporium species, we demonstrated that S. dehoogii displayed the best capacity to metabolize DCF when this latter is present in the medium as the sole carbon source. This observation led us to experiment the DCF biodegradation potential of our bio-electrochemical system composed of the improved CPE and S. dehoogii. This allowed us to observe a kinetic order of 1, a kinetic constant k (25°C) of 0.012 day-1 and a half time of 57.8 days, for an initial concentration of 1.65 ± 0.05 mg L-1. Further studies are undertaken to improve this process by using nanofibers cellulose (NFC) in order to increase the surface area and thus potentially to increase the CPE-Ce sensitivity. We will also test the possibility to associate the electrochemical analysis to other analytical methods (i.e. LC/MS) in order to decipher the catabolic pathway of DCF in S. dehoogii.
Acknowledgements
The authors wish to thank Romain Mallet (SCIAM, Angers University, France) for FEG-SEM images of our CPEs, Thomas GUILLEMETTE (IRHS, Angers, France), Hervé GAILLARD (ENSC Poitiers, France) and Didier HAUCHARD (ENSC Rennes, France) for fruitful discussions. A special thanks to Yordina GOVINDEN (M.Sc. student in Angers University in 2014) for the first results obtained on DCF electro analysis in the team Group Analysis and Processes (GA&P).
References
- FAFEOHS, Expert committee on water, Pharmaceuticals and drinking water Health risk assessment associated with presence of pharmaceuticals in drinking water : general method and application to carbamazepine and danofloxacin, 2013.
- Bannwarth B, Acetaminophen or NSAIDs for the treatment of osteoarthritis. Res Clin Rheumatol. 2006;20:117-29.
- H.A.T. directive, directive 2013/39/eu of the european parliament and of the council of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy, 2013.
- Westerhoff P, Yoon Y, Snyder S, et al. Fate of endocrine-disruptor, pharmaceutical, and personal care product chemicals during simulated drinking water treatment processes. Environ Sci Technol. 2005;39:6649-63.
- Kim SD, Cho J, Kim IS, et al. Occurrence and removal of pharmaceuticals and endocrine disruptors in South Korean surface, drinking, and waste waters. Water Res. 2007;41:10-13.
- Kruglova A, Ahlgren P, Korhonen N, et al. Biodegradation of ibuprofen, diclofenac and carbamazepine in nitrifying activated sludge under 12 °C temperature conditions. Sci Total Environ. 2014;409:394-401.
- Clauben M, Schmidt S. Biodegradation of phenol and p-cresol by the hyphomycete Scedosporium apiospermum. Res Microbiol. 1998;149:399-406.
- Santos VL, Heilbuth NM, Braga DT, et al. Phenol degradation by a Graphium sp. FIB4 isolated from industrial effluents. J Basic Microbiol. 2003;43:238-48.
- Blasi B, Polynter C, Rudavsky T, et al. Pathogenic yet environmentally friendly? Black fungal candidates for bioremediation of pollutants. Geomicrobiol J. 2016;33:308-17.
- Rougeron A, Giraud S, Alastruey-Izquierdo A, et al. Ecology of Scedosporium species: present knowledge and future research. Mycopathologia. 2018;183:185-200.
- Cortez KJ, Karoll J, Roilides E, et al. Infections caused by Scedosporium spp. Clin Microb Reviews. 2008;21:157-97.
- Sakata Y, Taga F, Ushigami T, et al. A Case of cutaneous mycosis caused by Scedosporium dehoogii on an immunocompromised patient. Mycopathologia. 2018;183:465-70.
- Mbokou SF, Pontié M, Razafimandimby B, et al. Evaluation of the degradation of acetaminophen by the filamentous fungus Scedosporium dehoogii using carbon-based modified electrodes. Anal Bioanal Chem. 2016;408:5895-903.
- Svancara Y, Kalcher K, Walcarius A, et al. Electrochemistry with carbon paste electrodes. CRS Press. 2012;666.
- Mokhtari A, Karimi-Maleh H, Ensafi AA, et al. Application of modified multiwall carbon nanotubes paste electrode for simultaneous voltammetric determination of morphine and diclofenac in biological and pharmaceutical samples. Sensors Actu B. 2012;169:96-05.
- Rajendra G, Sanghamitra C, Agrawal B. Electrochemical investigations of diclofenac at edge plane pyrolytic graphite electrode and its determination in human urine. Revue. 2010;145(2):743-8.
- Aguilar-Lira GY, Alvarez-Romero GA, Zamora-Suarez A, et al. New insight on diclofenac electrochemistry using graphite as working electrode. J Electroanal Chem. 2017;794:183-88.
- Afkhami A, Bahiraei A, Madrakian T. Gold nanoparticule/multiwalled carbon nanotube modified glassy carbon electrode as a sensitive voltammetric sensor for the determination of diclofenac sodium. Mat Sci and Eng C. 2016;59:168-76.
- Yang X, Wang F, Hu S. Enhanced oxidation of diclofenac sodium at a nano-structured electrochemical sensing film constructed by multi-wall carbon nanotubes-surfactant composite. Mat Sci Eng C. 2008;28:188-194.
- Gautam V, Singh KP, Yadav VL. Preparation and characterization of green-nano-composite material based on polyaniline, multiwalled carbon nano tubes and carboxymethyl cellulose: For electrochemical sensor applications. Carbohydr Polym. 2018;189:218-28.
- Ghalkhani M, Shahrokhian S. Development of a Nanocellulose Composite Based Voltammetric Sensor for Vitamin B9 Analysis. Curr nanosci. 2016;12(4):493-99.
- Mbokou SF, Pontié M, Bouchara JP, et al. Electrochemical performance of a carbon paste electrode modified by coffee husks for the quantification of acetaminophen in quality control of commercialized pharmaceutical tablets. Intern J Electrochem. 2016(2016):1-10.
- Pontié M, Mbokou SF, Bouchara JP, et al. Paracetamol sensitive cellulose-based electrochemical sensors. J Renew Mat, 2018;3:242-50.
- Pham T, Giraud S, Schuliar G, et al. Scedo-Select III: a new semi-selective culture medium for detection of the Scedosporium apiospermum species complex. Med. Mycol. 2015;53(5):512-19.
- Bard AJ, Faulkner IR. Electrochemical Methods: Fundamentals and Applications. John Wiley & Sons, New York, USA, 2nd Edition. 2001.
- Sbai S, Essis-Tome H, Gombert U, et al. Electrochemical stripping analysis of MPT using carbon fiber microelectrodes modified with combinations of poly-NiTSPc and Nafion films. Sens Actuators B Chem. 2007;124:368-75.
- Shalauddin M, Akhter S, Bagheri S, et al. Immobilized copper ions on MWCNTs-Chitosan thin film: enhanced amperometric sensor for electrochemical determination of diclofenac sodium in aqueous solution. Int J Hydrogen Energy. 2017;42:19951-60.
- Arvand M, Gholizadeh TM, Zanjanchi MA. MWCNTs/Cu(OH)2 nanoparticles/II. Nanocomposite modified glassy carbon electrode as a voltammetric sensor for the determination of the non-steroidal anti-inflammatory drug diclofenac. Mater Sci Eng C. 2012;32:1682-9.
- El-Wekil MM, Alkahtani SA, Refat H, et al. Advanced sensing nanomaterials based carbon paste electrode for simultaneous electrochemical measurement of esomeprazole and diclofenac sodium in human serum and urine samples. J Mol Liquids. 2018;262:495-503.
- Domaradza D, Guzik U, Wojcieszynska D. Biodegradation and biotransformation of polycyclic non-seroïdal anti-inflammatory drugs. Rev. Environ. Sci. Biotech. 2015;14:229-39.
- Facey SJ, Nebel BA, Kontny L, et al. Rapid and complete degradation of diclofenac by native soil microorganisms. Environ Technol Innov. 2018;10:55-61.