Original Article
, Volume: 12( 1)Diminutive Effects of Non-Covalent Interactions in Co...HClo4...H2O Triads
- *Correspondence:
- Abedien Z Department of Chemistry, Faculty of Science, Lorestan University, Khorramabad, Iran
Tel: +989161610549; E-mail: zebardasti@yahoo.com
Received: December 20, 2016 Accepted: January 26, 2017 Published: January 31, 2017
Citation: Zabardasti A, Hosseini SM. Diminutive Effects Of Non-Covalent Interactions in CO···HClO44···H2O Triads. Inorg Chem Ind J. 2017;12(1):105.
Abstract
Intermolecular interactions of different configurations in the HClO3···CO and HOClO3···H2O dyads, as well as CO···HClO3···H2O triad systems have been studied at MP2/6-311++G(2d,2p) computational level. Molecular geometries, binding energies, cooperative energies, many-body interaction energies and energy decomposition analysis (EDA) were evaluated. The results reveal that the stability of cyclic triads are more than linear and in the order IV > III > II > I configurations. All of the triads have diminutive energy. Red shifts of H-O stretching frequencies for complexes involving HOClO4 as H-donor are predicted. The electronic properties of the complexes are analyzed using parameters derived from the quantum theory of atoms in molecules (QTAIM) methodology.
References
Best Betting Sites in China Blog | Real Estate in Australia Blog | Real Estate in Spain Find Lawyer in Illinois Find Lawyer in Colorado Blog | Real Estate in France Info Alt Coins Reverse Mortgage in USA Blog | Real Estate in Japan Blog | Real Estate in Mexico Find Lawyer in Alabama Blog | Real Estate in Russia Car Insurance in Ohio Blog | Real Estate in Thailand Auto Insurance in New York Blog | Real Estate in Turkey Mortgage Calculator Blog | Mortgage Broker in USA Blog | Mortgage Company in USA Mortgage info in WashingtonKeywords
Intermolecular interactions; Cooperative effect; Diminutive effect; Many-body interaction energy; EDA; QTAIM
Introduction
Noncovalent interactions between molecules play a very important role in materials science, supramolecular chemistry and molecular biology [1-5]. The conventional hydrogen bonds (HB) as a noncovalent interaction A-H…B that involve electronegative atoms like oxygen and nitrogen have been thoroughly studied over the decades by experimental methods [1,2]. The chemical phenomenon of hydrogen bonding has also been studied extensively by quantum mechanical ab initio calculations [6-10]. Along with increasing of interest to hydrogen bonds, halogen bonds [11-13] are also taken into consideration. There have been numerous experimental and theoretical studies about the practical and potential applications of halogen bonds in different fields of biochemistry and supramolecular chemistry [14-22].
It has been suggested that chlorine resulting from the photo dissociation of chlorofluorocarbon in the stratosphere is involved in ozone-depleting catalytic cycles [23]. So the attentions were focused on the identifying of important reactions in the stratosphere that involve chlorine species [23]. Others said that, [24] sink role of perchloric acid (HOClO3) for stratospheric chlorine is more important than HCl. On the other hand CO and H2O are of primary importance in atmospheric chemistry[24,25]. So, we found it appropriate to examine interactions of CO and H2O with HOClO3, theoretically. Hence the present study specified to the analysis of structural parameters, vibrational stretching modes and electronic properties of intermolecular model systems formed by CO, H2O and HOClO3. In the other hand, triad structures including CO and H2O because of large difference between their dipole moments (CO experimentally about 0.12 D [26] pointing from carbon to oxygen and H2O 1.86 D [27,28] from hydrogen to oxygen) is an interesting design. On the best of our knowledge no research exists addressing the cooperative effect and many-body interaction analysis for these triads in the all probable configurations.
Method of Calculation
All of the calculations were performed using GAMESS package [29]. The geometries of the isolated HOClO3, CO and H2O moieties and their complexes were fully optimized at the MP2 computational levels with the 6-311++G(2d,2p) basis set [30]. Frequency calculations were performed at the same computational level in order to confirm that the structures obtained correspond to energy minima. BSSE correction was done by the counterpoise method as the most common way [31].
The AIM2000 package [32] was used to obtain bond electronic properties. The atoms in molecules (AIM) theory [32,33] was applied here to analyze the characteristics of the bond critical points (BCP) appearing in the studied systems.
Result and Discussion
Structure and stretching frequencies
The optimization structures of different 1:1:1 triad complexes from the association of HOClO3 with H2O and CO by forming the complexes (I), (II), (III) and (IV) are illustrated in Figure. 1.
Figure 1: Optimized geometries of the studied triads in all probably configurations and respective dimers.
To understand the properties of the systems better, the corresponding dyads are also studied.
In Table 1 the frequency shifts of the O-H stretching vibration in the dyads and triads relative to those in the isolated HOClO33 molecule are gathered. As frequency values show hydrogen-bond formation has been associated to a red shift in the stretching frequency of OH in HOClO3 as HB donor [34].
|
Configuration |
Complex | |||||
|---|---|---|---|---|---|---|
| HOClO3···CO | HOClO3···H2O | CO···HOClO3···H2O | ||||
| Δνstr | Δr | Δνstr | Δr | Δνstr | Δr | |
| I | -10.28 | 0.0008 | 2.38 | -0.0002 | -9.46 | 0.0008 |
| II | 1.59 | -0.0002 | -472.96 | 0.0238 | -450.77 | 0.0228 |
| III | -10.02 | 0.00081 | -473.32 | 0.0240 | -518.47 | 0.0263 |
| IV | -159.29 | 0.0078 | -473.32 | 0.0240 | -606.24 | 0.0308 |
?str and r of OH in isolated HOClO3 is 3736.43 cm-1 and 0.97152 Å respectively.
Table 1. Variation of bond stretching frequencies (Δνstr, cm-1) and bond length (Δr, Å) of O-H bond upon complexation.
An excellent linear correlation [Eq. (1)] was found between the frequency shifts and interaction distances as shown in Figure. 2. Furthermore a good linear relationship [Eq. (2)] was found between red shifts of O-H stretching vibrational frequency and density of the intermolecular BCP in HB complexes in Figure. 3.
Figure 2: Correlation between stretching frequency shifts and distances in all interactions of all studied complexes.
Figure 3: Correlation between densities of the intermolecular BCP in HB complexes and stretching frequency red shifts in O-H bond.
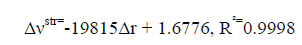 (1)
(1)
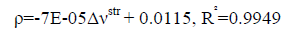 (2)
(2)
Energy analysis of the complexes
The values of the corrected stabilization energies ΔEC, (ΔE uncorrected=E supermolecule – E isolated monomers, ΔEC=ΔE uncorrected + BSSE) are listed in Table 2.
| Complex (A···B···C) CO···HOClO 3···H3O |
ΔEAB | ΔEBC | ΔEAC | ΔEABC | ΔE*AC | Ecoop |
|---|---|---|---|---|---|---|
| Configuration I | -5.73 | -5.29 | - | -11.00 | -0.10 | 0.12 |
| Configuration II | -2.65 | -41.17 | - | -42.77 | -0.12 | 1.17 |
| Configuration III | -5.72 | -41.16 | -2.33 | -46.60 | - | 2.62 |
| Configuration IV | -17.15 | -41.16 | -6.63 | -55.11 | - | 9.83 |
ΔE*: The values obtained from dyads frozen in the geometry of the triads.
Table. 2. Calculated stabilization energy ΔECint (kJ mol-1) and cooperative energy values Ecoop in the studied dyads and triads complexes.
The energy results reveal that the stabilization of complexes is in the order IV > III > II > I. This shows that the stability of cyclic are more than linear triads and the most stable complex is correspond to configuration IV. This difference in energy values depends on the interaction site of the CO donor species, O side or C side, and monomer positions.
An energetic cooperativity parameter was calculated using Eq. (3) and Eq. (4) for linear and cyclic triads respectively.
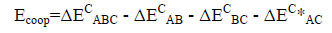 (3)
(3)
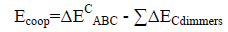 (4)
(4)
Where ΔEC*AC term is the interaction energy of AC dimer frozen in the geometry of the triad.
In the studied CO···HOClO3···H2O complex, diminutive effects are observed in all configurations with values as shown in Table 2. As shown as results no significant correlation found between the cooperative values and stabilities. Indeed the cooperativity and anti-cooperativity effects are mainly attributed to the charge transfer [35].
Many-body interaction analysis
The results in Table 2 show that CO···HOClO3···H2O in geometry IV and geometry I have maximum and minimum stabilization energy among studied complexes respectively.
As seen in Table 3 the main contribution of stabilization energy except the cases of geometry I is obtained by the two-body interaction ΔE2 B-C. This means that the OClO3H:OH2 interaction contributes more to the bonding interaction between pair of molecules in a triad than other interactions.
| CO···HOClO3···H2O (A···B···C) | ΔE2A-B | ΔE2B_C | ΔE2A-C | ΔE2A-B-C | ESS |
|---|---|---|---|---|---|
| Configuration I | -5.69 | -5.34 | -0.10 | -0.01 | 0.14 |
| Configuration II | 0.97 | -44.40 | -0.12 | -2.47 | 3.25 |
| Configuration III | -1.80 | -45.43 | -1.47 | -2.27 | 4.38 |
| Configuration IV | -3.97 | -46.04 | -5.11 | -5.62 | 5.62 |
Table 3. Decomposition of stabilization energy [kJ mol-1] of the studied triads using the geometry within the triads.
In the all studied systems the two-body 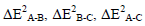 and three-body interactions ΔE3A-B-C have negative (attractive) values. The only case of two-body with positive value is ΔE2 HO4Cl-CO in configuration II that is contribute to HO4Cl: CO interaction.
and three-body interactions ΔE3A-B-C have negative (attractive) values. The only case of two-body with positive value is ΔE2 HO4Cl-CO in configuration II that is contribute to HO4Cl: CO interaction.
The strain energy (ES) is described as the energy sum of the monomers frozen in the geometry of the triads minus the energy sum of the isolated optimized monomers. So, the total binding energy of the triad is obtained using Eq. (5):
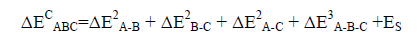 (5)
(5)
The strain energy (ES) can be defined as a measure of the degree of strain that drives the distortion of the ternary system. Equation (6) describes how calculate the ES. As seen in Eq. (6), ES calculated by sum of the monomers energies frozen in the geometry of the triads minus the energy sum of the optimized monomers.
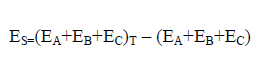 (6)
(6)
According to values in Table 3, the strain energy is positive, that is it makes a destabilizing contribution to the total stabilization energy of the triads.
Energy decomposition analysis
The energy-decomposition analysis (EDA) was performed to obtain insight into the source of the interaction energy using Eq. (7) [36].
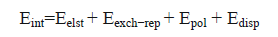 (7)
(7)
Where 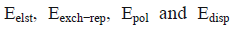 corresponds to electrostatic, exchange-repulsion, polarization and dispersion terms, respectively.
corresponds to electrostatic, exchange-repulsion, polarization and dispersion terms, respectively.
Table 4 lists the energy decomposition analysis results for perchloric acid complexes. The results reveal that electrostatic effects make the major contribution to the interaction energies (48.19% to 74.33%). This reveals that the electrostatic interactions are essentially responsible for the stability of the all studied complexes. Based on the energy decomposition results for dimers it is apparent that the polarization component in complexes with presence of hydrogen bonding interactions represent more values in comparison with the rest complexes. Also the electrostatic term for CO···HOClO3 dimers show larger values in configurations with oxygen interaction side (configurations I and III) of the CO than carbon interaction side (Configurations II and IV).
| Complex | Eelst | Eexch-rep | Epol | Edisp | EMP2 |
|---|---|---|---|---|---|
| CO···HOClO3 (I) | -12.68 (71.32%) |
11.05 | -5.10 (28.68%) |
0.92 | -5.82 |
| HOClO3···H2O (I) | -14.18 (74.01%) |
13.64 | -1.72 (8.98%) |
-3.26 (17.01%) |
-5.48 |
| CO···HOClO3···H2O (I) | -25.31 (74.33%) |
22.93 | -6.36 (18.68%) |
-2.38 (6.99%) |
-11.13 |
| CO···HOClO3 (II) | -6.57 (54.93%) |
9.25 | -0.75 (6.27%) |
-4.64 (38.80%) |
-2.68 |
| HOClO3···H2O (II) | -82.26 (69.40%) |
73.51 | -35.06 (29.58%) |
-1.21 (1.02%) |
-45.02 |
| CO···HOClO3···H2O (II) | -86.69 (68.61%) |
80.37 | -34.48 (27.29%) |
-5.19 (4.11%) |
-46.02 |
| CO···HOClO3 (III) | -12.59 (71.66%) |
10.92 | -4.98 (28.34%) |
0.88 | -5.77 |
| HOClO3···H3O (III, IV) | -82.42 (69.37%) |
73.81 | -35.23 (29.65%) |
-1.17 (0.98%) |
-45.02 |
| CO···HOClO3 (IV) | -24.52 (48.19%) |
33.01 | -14.56 (28.62%) |
-11.80 (23.19%) |
-17.87 |
| CO···HOClO3···H2O (IV) | -106.57 (61.69%) |
111.96 | -53.01 (30.68%) |
-13.18 (7.63%) |
-60.75 |
Table 4. EDA of perchloric acid complexes in kJ mol-1.
Electron density analysis
In Table 5, are listed the results of the QTAIM topological parameters, namely as electronic density (ρ), Laplacian (∇2) and the ratios between the kinetic (G) and potential (V) electron energy.
| Complex | Configuration | Interaction | Interaction distance | ρ | ∇2 | -G/V |
|---|---|---|---|---|---|---|
| HOClO3···CO | I | OClO3H:OC | 2.17 | 0.0123 | 0.0509 | 1.22 |
| HOClO3···H2O | I | HO4Cl:OH2 | 3.24 | 0.0075 | 0.0328 | 1.24 |
| CO···HOClO3···H2O | I | O3ClOH:OC | 2.19 | 0.0119 | 0.0490 | 1.23 |
| CO···HOClO3···H3O | I | HClO3O:OH2 | 3.21 | 0.0067 | 0.0264 | 1.22 |
| CO···HOClO3···H2O | I | HClO3O: OH2 | 3.21 | 0.0066 | 0.0266 | 1.24 |
| HOClO3···CO | II | HO4Cl:CO | 3.47 | 0.0045 | 0.0224 | 1.59 |
| HOClO3···H2O | II | O3ClOH:OH3 | 1.70 | 0.0411 | 0.1319 | 0.94 |
| CO···HOClO3···H2O | II | HO4Cl :CO | 3.52 | 0.0046 | 0.0209 | 1.59 |
| CO···HOClO3···H2O | II | O3ClOH :OH2 | 1.71 | 0.0433 | 0.1212 | 0.89 |
| HOClO3···CO | III | O3ClOH:OC | 2.18 | 0.0121 | 0.0500 | 1.22 |
| HOClO3···H2O CO···HOClO 3···H2O |
III, IV III |
O3ClOH:OH2 O 3ClOH:OH2 |
1.70 1.68 |
0.0413 0.0465 |
0.1323 0.1257 |
0.94 0.87 |
| CO···HOClO3···H2O | III | CO:HOH | 2.19 | 0.0109 | 0.0469 | 1.26 |
| CO···HOClO3···H2O | III | HClO3O:OC | 3.13 | 0.0049 | 0.0186 | 1.15 |
| HOClO3···CO CO···HOClO 3···H2O |
IV IV |
O3ClOH:CO OClO 3H:OH2 |
2.09 1.65 |
0.0224 0.0504 |
0.0600 0.1300 |
1.00 0.85 |
| CO···HOClO3···H2O | IV | OC:HOH | 2.17 | 0.0180 | 0.0539 | 1.09 |
| CO···HOClO3···H2O | IV | HClO3O:CO | 3.10 | 0.0062 | 0.0209 | 1.23 |
Values of ρ and ∇2 ρ are given in e.ao-3 and e.ao-5, respectively
Table 5. Computed values of the QTAIM topological parameters at the BCP.
The topological analysis of the electron density displays the presence of an intermolecular bond critical point (BCP) in all of the complexes, HB and vdW.
In all cases, these BCPs show small values of the electron density and small positive Laplacians, an indication of the closed-shell characteristics of the interaction, similar to those found in weak interactions [37-40]. The –G/V value is indicating interaction nature as covalent or non-covalent [41]. The –G/V values are higher than 1, between 1 and 0.5 and lower than 0.5 indicating interaction with non-covalent, partial covalent and covalent characteristics, respectively. So the values in Table 5 illustrate that the OClO3H:OC and O3ClOH:OH2 interactions are interactions with non-covalent and partial covalent characteristics, respectively.
As shown as Figure. 4 and Figure. 5 there is good linear correlation between –G/V values and interaction distances. Linear correlation between –G/V values with OClO3H:OC and O3ClOH:OH2 interaction distances displaying by Eq. (8) and Figure. 4. Also Figure. 5 and Eq. (9) explain linear correlation between –G/V values and O:O, O:Cl and Cl:C intermolecular interaction distances.
Figure 4: Correlation between ?G/V with OClO3H···OC and O3ClOH···OH2 interaction distances.
Figure 5: Correlation between ?G/V with O:O, O:Cl and Cl:C intermolecular interaction distances.
-G/V=0.6642 r(H···O) - 0.2237, R²=0.9811 (8)
-G/V=1.0927 r - 2.2486, R²=0.9241 (9)
The electron density in BCP represents the strength of a bond. Generally, larger values of ρ show a stronger bond [42]. So according to the values, the strength of HB interactions is larger than vdW interactions.
Conclusions
Theoretical study of the 1:1 HOClO3···CO and HOClO3···H2O dyad complexes as well as 1:1:1 H2O···HOClO3···CO triad complex were investigated by quantum chemical calculations at the MP2/6-311++G(2d,2p) level. The triad systems were located in 4 configurations, 2 linear and 2 cyclic.
The vibrational analysis of the studied complexes revealed that HB formation has been associated to a red shift in the stretching frequency of the O-H in perchloric acid molecule.
Many body analyzes indicated that the HOClO3···H2O interaction contributes more to the bonding interaction between two molecules in a triad than other interactions.
Based on the energy decomposition analysis (EDA), it can be seen that the electrostatic interactions are essentially responsible for the stability of the all complexes.
Good linear correlation was obtained from AIM results between –G/V and interaction distances. We think that the results of present study may be useful for understanding competitive role of carbon monoxide, perchloric acid and water molecules in atmospheric chemistry.
References
- Desiraju, G, Steiner T. The weak hydrogen bond. Oxford Univ. Press, 1999.
- Jeffrey GA. Jeffrey GA. An introduction to hydrogen bonding. Oxford University press New York, 1997.
- Scheiner, S. Molecular Interactions. From van der waals to strongly bound complexes. John Wiley & Sons, Chichester, UK, 1997.
- Gilli G, Gilli P. The nature of the hydrogen bond: outline of a comprehensive hydrogen bond theory. OUP Oxford, 2009.
- Grabowski SJ. (Ed.) Hydrogen bonding: New insights, Springer, 2006.
- Blanco F, Alkorta I, Solimannejad M, et al. Theoretical study of the 1:1 complexes between carbon monoxide and hypohalous acids J. Phys. Chem. A. 2009;113(13)3237-44.
- Zabardasti A, Kakanejadifard A, Kikhaei M, et al. Theoretical studies and topological analysis of the electron density of clusters of O3 with HNCO and HCNO, Comp. Theor. Chem. 2010;961,1-5.
- Zabardasti A, Kakanejadifard A. Theoretical study of hydrogen bonded clusters of water and cyanic acid: Hydrogen bonding in terms of the molecular structure, Polyhedron. 2008;27(13),2973-77.
- Zhao GM, Liu YC, Shi WJ, et al. A theoretical investigation into the cooperativity effect involving anionic hydrogen bond, thermodynamic property and aromaticity in Cl−···benzonitrile···H2O ternary complex. Comp. Theor. Chem. 2014;1035, 76-83.
- Zabardasti A, Solimannejad M. Theoretical study of hydrogen bonded clusters of water and fulminic acid, Comp. Theor. Chem. 2007;810(1),73-9.
- Jab?o?ski M, Palusiak M. Nature of a hydride–halogen bond. A SAPT-, QTAIM-, and NBO-Based Study, J. Phys. Chem. A. 2012;116(9)2322-32.
- Lipkowski P, Grabowski SJ, Leszczynski J, et al. Properties of the halogen−hydride interaction: An ab initio and “Atoms in molecules” analysis. J. Phys. Chem. A. 2006;110(34)10296-302.
- Solimannejad M, Hosseini SM, Zabardasti A. Cooperative and diminutive interplay between halogen, hydride and cation-σ interactions. Phys. Chem. Res. 2016;4(4)583-589.
- Solimannejad M, Rezaei Z, Esrafili MD, Interplay and competition between the lithium bonding and halogen bonding: R3C···XCN···LiCN and R3C···LiCN···XCN as a working model (R=H, CH3; X=Cl, Br). Mol. Phys. 2014;112(13):1783 -1788.
- Auffinger P, Hays FA, Westhof E, et al. Halogen bonds in biological molecules. Acad. Sci. USA. 2004;101(48),16789- 94.
- Hardegger LA, KuhnL A, Spinnler B, et al. Systematic investigation of halogen bonding in protein–ligand interactions. Angew. Chem. Int. Ed. 2011;50(1)314-318.
- Metrangolo P, Resnati G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001;7(12), 2511-19.
- Solimannejad M, Orojloo M, Amani S. Effect of cooperativity in lithium bonding on the strength of halogen bonding and tetrel bonding: (LiCN)n···ClYF3 and (LiCN)n···YF3Cl (Y=C, Si and n = 1-5) complexes as a working model. J. Mol. Model. 2015;21(7),183.
- Solimannejad M, Bayatmanesh E, Esrafili MD. Interplay between lithium bonding and halogen bonding in F3CX•••YLi•••NCCN and F3CX•••NCCN•••LiY complexes (X=Cl, Br; Y=CN, NC), Phys. Chem. Res. 2014;2,171-78.
- Solimannejad M, Malekani M, Alkorta I. Substituent effects on the cooperativity of halogen bonding, J. Phys. Chem. A. 2013, 117(26),5551-57.
- Solimannejad M, Malekani M. Cooperative and diminutive interplay between the hydrogen bonding and halogen bonding in ternary complexes of HCCX (X=Cl, Br) with HCN and HNC, J. Phys. Chem. A. 2012;998,34-38.
- Solimannejad M, Malekani M, Alkorta I. Cooperativity between the hydrogen bonding and halogen bonding in F3CX ··· NCH(CNH) ··· NCH(CNH) complexes (X=Cl, Br), Mol. Phys. 2011;109(13)1641-48.
- Molina MJ, Rowland FS. Stratospheric sink for chlorofluoromethanes: Chlorine atom-catalysed destruction of ozone. Nature. 1974; 249,810-12.
- Francisco JS. Ab initio characterization of HOClO3 and HO4Cl: Implications for atmospheric chemistry. J. Phys. Chem. 1995;99(36)13422-25.
- Simonaitis R, Heicklen J. Perchloric acid: A possible sink for stratospheric chlorine. Planet. Space Sci, 1975;23(11)1567-69.
- Harrison JF. Relationship between the charge distribution and dipole moment functions of CO and the related molecules CS, SiO, and SiS. J. Phys. Chem. A. 2006;110(37)10848-57.
- Dyer PJ, Cummings PT. Hydrogen bonding and induced dipole moments in water: Predictions from the Gaussian charge polarizable model and Car-Parrinello molecular dynamics. J. Chem. Phys. 2006;125(14)144519.
- Bucher D, Kuyucak S. Polarization of water in the first hydration shell of K+ and Ca2+ ions. J. Phys. Chem. B. 2008;112(35),10786-90.
- Schmidt MW, Baldridge KK, Boatz JA, et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14(11)1347-63.
- Frisch MJ, Pople JA, Binkley J. Self-consistent molecular orbital methods: Supplementary functions for Gaussian basis sets, J. Chem. Phys. 1984;80(7)3265.
- Boys SF, Bernardi FD. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19(4)553-566.
- Bader RFW. Atoms in molecules: A quantum theory. Oxford University Press, Oxford, 1990.
- Popelier PLA. Atoms in molecules, An introduction, Pearson Education Limited, Prentice Hall, 2000.
- Gu QY, Lou SC. Table of chemical materials, Jiangsu Science and Technology Press: Jiangsu, China, 1998.
- Glendening ED, Natural energy decomposition analysis: Extension to density functional methods and analysis of co-operative effects in water clusters. J. Phys. Chem. A. 2005, 109(51)11936-40.
- Su P, Li H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009;131(1)014102.
- Alkorta I, Elguero J, Non-conventional hydrogen bonds. J. Chem. Soc. ReV. 1998, 27(2)163-170.
- Bone RGA, Bader RFW. Identifying and analyzing intermolecular bonding interactions in van der waals molecules, J. Phys. Chem. A. 1996;100(26),10892-10911.
- Rozas I, Alkorta I, Elguero J. Behavior of ylides containing N, O, and C atoms as hydrogen bond acceptors. J. Am. Chem. Soc. 2000;122(45)11154-61.
- Zabardasti A, Goudarziafshar H, Salehnassaj M, et al. computational study of hydrogen bonds in intermolecular systems of high complexity: arachno-pentaborane(11)···Y with Y = O2 and N2, J. Mol. Model. 2014; 20(9), 2403.
- Grabowski SJ. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111(4) 2597-625.
- Matta CF, Boyd RJ. Quantum theory of atoms in molecules: From solid state to DNA and drug design, 2007; Ch.1, pp.134.