Original Article
, Volume: 10( 3)Utility of 2-Cyano-3, 3-Diphenylprop-2-enoyl Isothiocyanate as a Source of Quinazoline, Pyrimidine and Oxazine Derivatives
- *Correspondence:
- Hemdan MM, Department of Chemistry, Faculty of Science, Ain Shams University, 11566, Abbasia, Cairo, Egypt, Tel: +20 2 26831474; E-mail: mhemdan39@hotmail.com
Received: September 08, 2017; Accepted: October 09, 2017; Published: October 11, 2017
Citation: Hemdan MM, Fahmy AF, Nawal FA, et al. Utility of 2-Cyano-3, 3-Diphenylprop-2-Enoyl Isothiocyanate as a Source of Quinazoline, Pyrimidine and Oxazine Derivatives. ChemXpress. 2017;10(3):129
Abstract
2-Cyano-3, 3-diphenylprop-2-enoyl isothiocyanate (1) is used as a building block in synthesis of of quinazoline, pyrimidine and oxazine derivatives. The electron impact ionization mass spectra of the synthesized compounds were studied. The MS peaks obtained correspond very well with their fragmentation patterns.
Keywords
Quinazoline; Pyrimidine; Oxazine derivatives; Mass spectroscopy
Introduction
Extensive studies on the chemistry of aroyl isothiocyanates have established the value of these reagents as starting materials for the synthesis of a wide variety of heterocyclic compounds and thiourea derivatives [1-11]. Highly reactive α, β-unsaturated acyl isothiocyanates can be useful intermediates in organic synthesis because their multiple bonds can participate in some cyclization reactions [12]. Intermolecular cyclization of α, β-unsaturated acylthioureas, formed in the reaction of α, β-unsaturated acyl isothiocyanates and amines were reported [13-16].
In the present work we attempted to activate the C=C bond by introduction of an electron-accepting substituent into the α-position of the 3, 3-diphenylprop-2-enoyl residue. We choose the cyano group as the electron-accepting substituent.
The structures of compounds 2-4 are substantiated from their microanalytical and spectral data. Thus, their IR spectra show bands attributable to NH, CO groups; in addition to CN group in case of compounds 2 and 3. Their 1HNMR spectra show aromatic protons as well as broad signals correspond to acidic NH and OH protons in the downfield region that exchangeable with D2O shake. Further highlights on the assigned structures of the synthesized compounds were gained from their mass spectra (MS) that revealed MS peaks correspond very well with their proposed structures (see Experimental). The formation of compound 2 can be visualized on the basis of nucleophilic attack of amino group of anthranilic acid on the electrophilic carbon atom of isothiocyanato group of compound 1. Compound 3 is formed via elimination of water molecule from thiourea derivative 2. On the other hand, formation of compound 4 is understood through cleavage of the amide linkage of cinnamoyl part of thiourea 2; followed by elimination of a molecule of water or vice versa.
Experimental
Melting points were determined in open capillary tubes on a Gallenkemp melting point apparatus and are uncorrected. The infrared spectra were recorded on Perkin-Elemer 2000 FTIR and Maston 1000-FTIR spectrometer. The 1HNMR spectra were measured on Jeol (EX-400) spectrometer with chemical shift (δ) expressed in ppm downfield from TMS as internal standard, in DMSO-d6 or CDCl3. All acidic protons disappeared by deuterium exchange (addition of D2O). Mass spectra were determined with Jeol-JMS, DX 303. Elemental analyses wer determined by CHN Carder-MT-3 Yanaco and Perkin-Elemer 2400 CHN, elemental analyzer. TLC was carried out the monitoring of the progress of all reactions and homogeneity of the synthesized compounds. TLC was determined using TLC aluminum sheets silica gel F254 (Merck).
Synthesis of 2-Cyano-3, 3-diphenylprop-2-enoyl isothiocyanate
To a solution of 2-cyano-3, 3-diphenylprop-2-enoyl chloride 16 (3 mmol) in dry acetone (30 mL), solid ammonium thiocyanate (3 mmol) was added. The reaction mixture was stirred for 30 min at room temperature. The precipitated ammonium chloride was filtered off to give a clear solution of isothiocyanate 1.
Reaction of isothiocyanate 1 with different nucleophiles
General procedure: To a solution of isothiocyanate l (3 mmol) in a dry acetone (50 mL), anthranilic acid was added. The reaction mixture was refluxed for 3 h and cooled to room temperature. The precipitated solid was filtered off and recrystallised from benzene/ethanol mixture to give compound 2. The same procedure was done with aniline, p-toludine, p-anisidine, benzoyl hydrazine, phenylhydrazine and/or water. The progress of all reactions and homogeneity of the synthesized compounds was monitored by TLC. The solid obtained for each reaction was crystalized from a suitable solvent to give the corresponding compounds.
2-(3-(2-Cyano-3, 3-diphenylacryloyl)thioureido) benzoic acid (2): 75% Yield; yellow crystals; m.p 183°C with decomposion, (benzene-ethanol mixture); IR (KBr) υ: 3408 (br. OH), 3254 (NH), 2240 (CN), 1710, 1688 (C=O), 1384 (CS); 1HNMR (DMSO-d6) δ: 7.25 (d, 2H, J=5.49 Hz), 7.36 (t, 1H, J=5.49 Hz), 7.43-7.61 (m, 9H, ArH), 7.94 (d, 1H, J=5.49 Hz), 8.11 (d, 1H, J=5.49 Hz), 11.79, 12.60, (two br. s, 2H, 2NH, exchangeable with D2O); 13.57 (br. s, 1H, OH, exchangeable with D2O); MS (70eV) m/z (%): 427 (M+., 0), 349 (5), 299 (10), 248 (20), 247 (10), 232 (100), 204 (25), 179 (20), 137 (20), 119 (40), 77(30); Anal. Calcd. for C24H17N3O3S (427.48): C, 67.43; H, 4.01; N, 9.83. Found C, 67.80; H, 3.94; N, 9.65%.
4-Oxo-1, 6, 6-triphenyl-2-thioxo-hexahydropyrimidine-5-carbonitrile (6a): 85% yield; yellow crystals; m.p 205-207°C, (light petroleum 60-80 – benzene mixture); IR (KBr) υ: 3245 (NH), 2220 (CN), 1670 (C=O), 1310 (CS); 1HNMR (CDCl3) δ: 3.8 (s, 1H, CH), 6.8-8.2 (m, 15H, ArH), 11.86, 12.96, (two br. s, 1H, NH, exchangeable with D2O); MS (70eV) m/z (%): 383 (M+., 40), 306(1), 264 (1), 248 (40), 247 (100), 232 (80), 204 (35), 136 (5), 135(75), 105 (20), 77(30); Anal. Calcd. for C23H17N3OS (383.47): C, 72.04; H, 4.47; N, 10.96. Found C, 72.42; H, 4.21; N, 10.46%.
4-Oxo-6, 6-diphenyl-2-thioxo-1-p-tolyl-hexahydropyrimidine-5-carbonitrile (6b): 80% yield; yellow crystals; m.p 203-205°C, (light petroleum 60-80 – benzene mixture); IR (KBr) υ: 3210 (NH), 2229 (CN), 1676(C=O), 1340 (CS); 1HNMR (CDCl3) δ: 2.3 (s, 3H, CH3), 3.7 (s, 1H, CH), 6.9-8.3 (m, 14H, ArH), 11.83, 12.00, (two br. s, 1H, NH, exchangeable with D2O); MS (70eV) m/z (%): (M+., 40); Anal. Calcd. for C24H19N3OS (397.49): C, 72.52; H, 4.82; N, 10.57. Found C, 72.89; H, 4.69; N, 10.33%.
1-(4-Methoxyphenyl)-4-oxo-6,6-diphenyl-2-thioxo hexahydro-pyrimidine-5-carbonitrile (6c): 80% yield; yellow crystals; m.p 185-187°C, (light petroleum 60-80 – benzene mixture); IR (KBr) υ: 3246 (NH), 2235 (CN), 1672(C=O), 1370 (CS); 1HNMR (CDCl3) δ: 3.49 (s, 3H, OCH3), 3.87 (s, 1H, CH), 6.76-8.13 (m, 14H, ArH), 10.62, 11.54, (two br. s, 1H, NH, exchangeable with D2O); Anal. Calcd. for C24H19N3O2S (413.49): C, 69.71; H, 4.63; N, 10.16. Found C, 70.08; H, 4.73; N, 9.98%.
N-(5-Cyano-4-oxo-6,6-diphenyl-2-thioxo-tetrahydropyrimidin-1(2H)-yl) benzamide (6d): 70% yield; yellow crystals; m.p 130-131°C, (benzene); IR (KBr) υ: 3238 (NH), 2228 (CN), 1680 (C=O), 1310 (CS); 1HNMR (CDCl3) δ: 3.8 (s, 1H, CH), 6.84 -7.94 (m, 15H, ArH), 8.67, 8.84, (two br. s, 1H, NH, exchangeable with D2O), 12.71 (br. s, 1H, NH, NHCOPh, exchangeable with D2O); MS (70eV) m/z (%): 426 (M+., 1), 348(8), 305(51), 299 (20), 248 (5), 247 (10), 232 (30), 205 (5), 177 (20), 127(5), 105 (20), 78(100); Anal. Calcd. for C24H18N4O2S (426.49): C, 67.59; H, 4.25; N, 13.14. Found C, 67.32; H, 4.31; N, 12.87%.
34-(2-Cyano-3,3-diphenylacryloyl)-2-phenyl-1-(propan-2-ylidene) thiosemicarbazide (7): 78% yield; yellow crystals; m.p 180-183°C, (benzene); IR (KBr) υ: 3210, (NH), 2220 (CN), 1680 (C=O), 1320 (CS); 1HNMR (CDCl3) δ: 0.87 (s, 6H, 2CH3), 6.58-7.34 (m, 15H, ArH), 9.11 (br. s, 1H, NH, NHCO, exchangeable with D2O); 13C NMR (CDCl3) 21.10 (CH3), 23.42 (CH3), 81(Cb), ar-C [124.68 (2CH), 126.57 (2CH), 127.33 (1CH), 128.32 (2CH), 128.76 (2CH), 129.57 (2CH), 130.19 (1CH), 130.81 (2CH), 131.39 (1CH), 138.18 (1C), 138.76 (1C), 139.04 (1C)], 117.81 (CN), 157 (Cc=N) 163.29 (Ca) 173.43 (CO), 185.76 (CS); MS (70eV) m/z (%): 438 (M+., 6), 437 (1), 423 (49), 232 (100), 206 (4), 204 (23), 177 (13), 148 (44), 105 (17), 92 (20); Anal. Calcd. for C26H22N4OS (438.54): C, 71.21; H, 5.06; N, 12.78;. Found C, 70.87; H, 4.71; N, 12.62%.
4-Oxo-6,6-diphenyl-2-thioxo-1,3-oxazinane-5-carbonitrile (9): 80% yield; yellow crystals; m.p 191-192°C, (benzene–ethanol mixture); IR (KBr) υ: 3261, 3192 (NH), 2228 (CN), 1695(C=O), 1328 (CS); 1HNMR (DMSO-d6) δ: 7.0-7.65 (m, 10H, ArH), 9.3, 9.5 (two s, 1H, CH), 11.0, 11.50, (two br. s, 1H, 1NH, exchangeable with D2O), 12.00 (br. s, 1H, 1OH, exchangeable with D2O); MS (70eV) m/z (%): 308 (M+., 15), 307(60), 248 (3), 247 (10), 232 (100), 204 (30), 203 (30), 177 (40), 105(30), 77(40); Anal. Calcd. for C17H12N2O2S (308.35): C, 66.22; H, 3.92; N, 9.08. Found C, 66.39; H, 4.25; N, 8.78%.
Heating of compound 2 with acetic anhydride
General procedure: A solution of compound 2 in acetic anhydride (20 mL) was heated at 90°C for 1h. A solid product was obtained after cooling which was filtered off and re-crystallised from benzene to give compound 3. The same procedure was done with heating of compound 2 for 3 h. to give compound 4.
2-(4-Oxo-2-thioxo-1, 2, 3, 4-tetrahydroquinazoline-3-carbonyl)-3, 3-diphenylacrylonitrile (3): 80% yield; yellow crystals; m.p 160-162°C, (benzene); IR (KBr) υ: 3200 (NH), 2210 (CN), 1680 (C=O), 1380 (CS); 1HNMR (DMSO-d6) δ: 7.25 (d, 1H, J=6.20 Hz), 7.33 (t, 1H, J=5.6 Hz), 7.41 (d, 1H, J=5.32 Hz), 7.46-7.69 (m, 9H, ArH), 7.72 (t, 1H, J=6.32 Hz), 7.96 (d, 1H, J=6.18 Hz), 10.45, (br. s, 1H, 1NH, exchangeable with D2O); MS (70eV) m/z (%): 409 (M+., 1), 381 (12), 375 (21), 299 (10), 246 (17), 232 (100), 204 (43), 177 (16), 133 (5), 77(30); Anal. Calcd. for C24H15N3O2S (409.46): C, 70.40; H, 3.69; N, 10.26; Found C, 70.21; H, 3.94; N, 10.10%.
2-Thioxo-2, 3-dihydroquinazolin-4(1H)-one (4): 75% yield; colorless crystals; m.p 270-271°C (benzene); IR (KBr) υ: 3210 (NH), 1695 (C=O), 1380 (CS); 1HNMR (DMSO-d6) δ: 7.32 (t, 1H, J=5.85 Hz), 7.35 (d, 1H, J=6.24 Hz), 7.72 (t, 1H, J=6.57 Hz), 7.92 (d, 1H, J=6.92 Hz), 12.50, 12.70, (two br. s, 2H, 2NH, exchangeable with D2O); MS (70eV) m/z (%): 178 (M+., 100), 150 (10), 145 (15), 120 (50), 119 (45), 92 (40), 65(5); Anal. Calcd. for C8H6N2OS (178.21): 53.92; H, 3.39; N, 15.72. Found C, 53.99; H, 3.33; N, 15.40%.
2-Cyano-3, 3-diphenylacrylamide (10): A solution of compound 6a (1 gm) in 20 mL glacial acetic acid was refluxed for 3 h. in the presence of fused sodium acetate (0.3 gm). The reaction mixture was filtrated while hot and then cooled to room temperature. A solid product is obtained, filtered, washed several times with hot water and then recrystallized from acetic acid to give compound 10. The same product was obtained in case of compound or 6b or 6c. 89% yield; colorless crystals; m.p 222-224°C; IR (KBr) υ: 3454, 3331, 3184 (NH), 2212 (CN), 1688 (C=O), 1371(CS); 1HNMR (DMSO-d6) δ: 7.16 (t, 2H, J=6.07 Hz), 7.26 (t, 2H, J=6.06 Hz), 7.28 (t, 2H, J=6.00 Hz), 7.34 (d, 2H, J=6.03), 7.38 (d, 2H, J=6.05), 11.22 (br. s, 2H, NH, exchangeable with D2O); MS (70eV) m/z (%): 248 (M+., 100), 247 (78), 232 (17), 204 (89), 178 (34), 165 (44), 77(42); Anal. Calcd. for C16H12N2O2 (248.28): C, 77.40; H, 4.87; N, 11.28. Found C, 77.32; H, 4.76; N, 10.96%.
Ethyl 2-cyano-3, 3-diphenylacrylate (11): A solution of compound 6a (1 gm) in 300 mL absolute ethanol was refluxed for 5 h in the presence of sodium acetate (0.3 gm). The reaction mixture was filtrated while hot and then cooled to room temperature. A solid product is obtained, filtered, washed several times with hot water, dried and then recrystallized from light petroleum ether 60°C-80°C to give compound 11. |The same product was obtained in case of compound or 6b or 6c. 79% yield; colorless crystals; m.p 105-107°C; IR (KBr) υ: 2220 (CN), 1742 (C=O); 1HNMR (DMSO-d6) δ: 1.55 (m, 3H, CH3CH2), 3.72 (m, 2H, CH3CH2), 7.16 (d, 2H, J=5.49 Hz), 7.36-7.57 (m, 8H, ArH); MS (70eV) m/z (%): 277 (M+., 12), 263 (100), 248 (16), 232 (81), 204 (76), 177 (35), 165 (13), 77(43); Anal. Calcd. for C18H15NO2 (277.32): C, 77.96; H, 5.45; N, 5.05. Found C, 78.23; H, 5.08; N, 4.98%.
Materials and Methods
The interaction of equimolar quantities of 2-cyano-3, 3-diphenylprop-2-enoyl isothiocyanate (1) with anthranilic acid in a dry acetone gave a good yield of linear 1:1 adduct 2. The thiourea derivative 2 proved less prone to cyclize under the reaction conditions even with its refluxing for long time in acetonitrile. However, acetic anhydride affected ring closure of 2 with elimination of a molecule of water to afford the quinazoline derivatives 3. Moreover, quinazoline 4 was obtained upon further heating of the adduct 2 in acetic anhydride for long time as shown in Scheme 1.
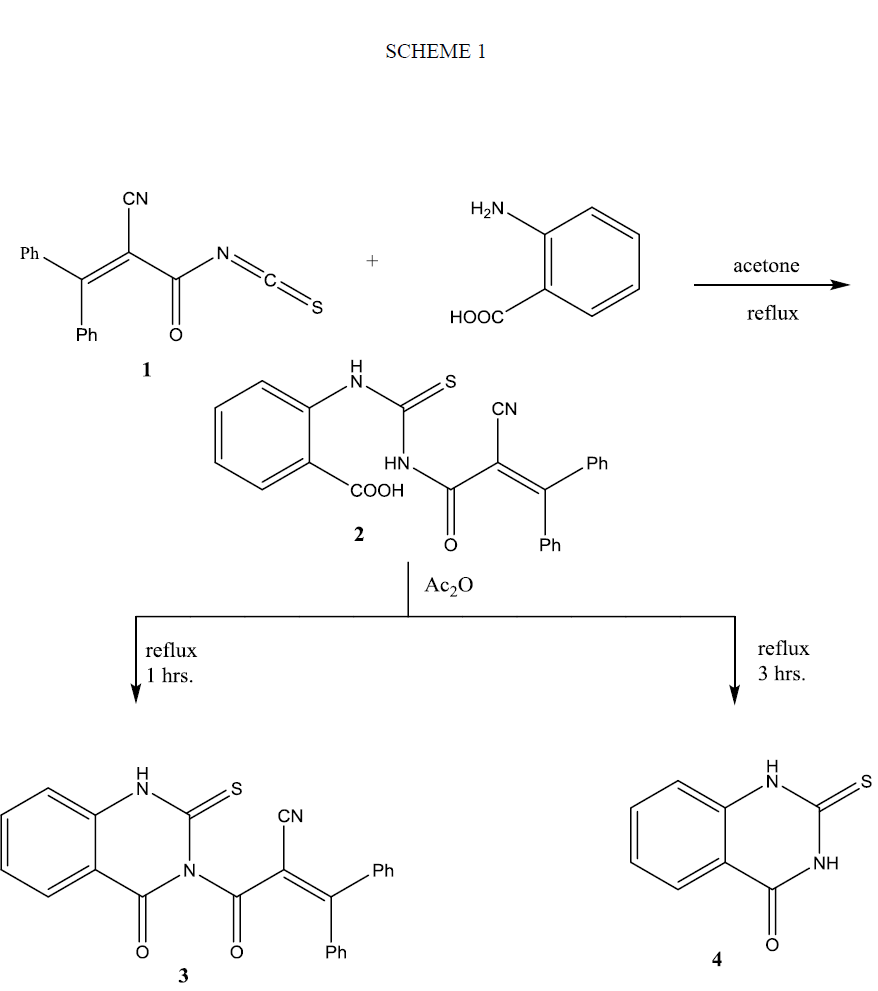
Treatment of a solution of isothiocyanate 1 in a dry acetone with aromatic amines such as aniline, p-toludine and/or p-anisidine not give the expected open adducts 5(a-c); instead it produced pyrimidine derivatives 6(a-c) in good yield as shown in Scheme 2. Similar treatment of 1 with benzoyl hydrazine furnished pyrimidine derivative 6d. On the other hand, the reaction of 1 with phenyl hydrazine gave thiosemicarbazide derivative 7. The structures of the synthesized compounds 6(a-d) and 7 were elucidated from their micro-analytical and spectral data that revealed a pattern completely in accord with their proposed structures. Thus, their IR spectra showed absorption bands correlated with υ (NH), υ (CN), υ (C=O) and in addition to υ (C=S). Their 1HNMR spectral data showed signals correspond to aromatic protons, as well as, NH protons in the downfield region that exchangeable with D2O shake, in addition to alkyl protons. Inspection the 1HNMR spectra of compounds 6(a-d) showed two broad signals of NH proton corresponds to one proton. This suggests the existence of compounds 6(a-d) as a mixture of two diastereo ismomers (see Experimental). 13C chemical shifts of compound 7 showed the different types of carbon atoms, including CO signal at 173.43 ppm and CS signal at 185.76 ppm and in addition to signals at 21.10 and 23.42 for methyl group’s carbon atoms. Further evidence on the assigned structures of compounds 6(a-d) and 7 is gained from their mass spectra that showed their correct molecular ion peak as well as important fragments that on accord with their structures. The formation of compounds 6(a-d) is explained on the basis of cyclization of the intermediate thiourea 5(a-d) (not isolated) via nucleophlic attack at the β-carbon atom of the unsaturated amide. The formation of compound 7 can be explained on the basis of nucleophilic addition of isopropylidene phenyl hydrazone (obtained as a side product of the reaction of phenyl hydrazine with acetone) on the carbon atom of isothiocyanato group.
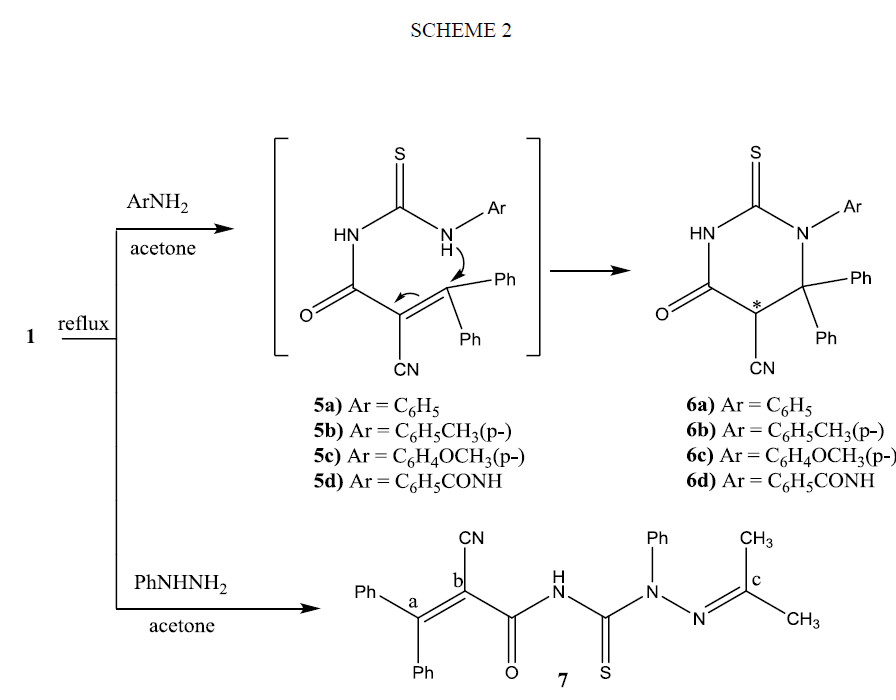
The heteroallene 1 reacts readily with water to yields the stable 4-oxo-6, 6-diphenyl-2-thioxo-1, 3-oxazinane-5-carbonitrile (9) in a good yield (Scheme 3). The structure assigned to compound 9 was based on microanalytical and spectral data. Its IR spectrum showed absorption bands correlated with OH, NH, CN and CO groups. The 1HNMR spectrum of 9 showed a multiplet signal at δ: 7.00 ppm to 7.65 ppm corresponds to 10 aromatic protons, two singlets at δ 9. 3 and 9.5 ppm integrating for one proton, correspond to the methine proton Ha. The proton Ha is highly de shielded by the neighboring carbonyl and cyano group as well as it affected by the anisotropic field of the two phenyl rings. Moreover, it reveals two brood signals correspond to one proton, in the downfield region at δ 11.0 and 11.50 ppm exchangeable with D2O for NH proton beside a signal at 12.00 ppm for OH proton. Further highlights on the assigned structure of compound 3 were given from its mass spectrum that revealed its correct molecular ion peak.
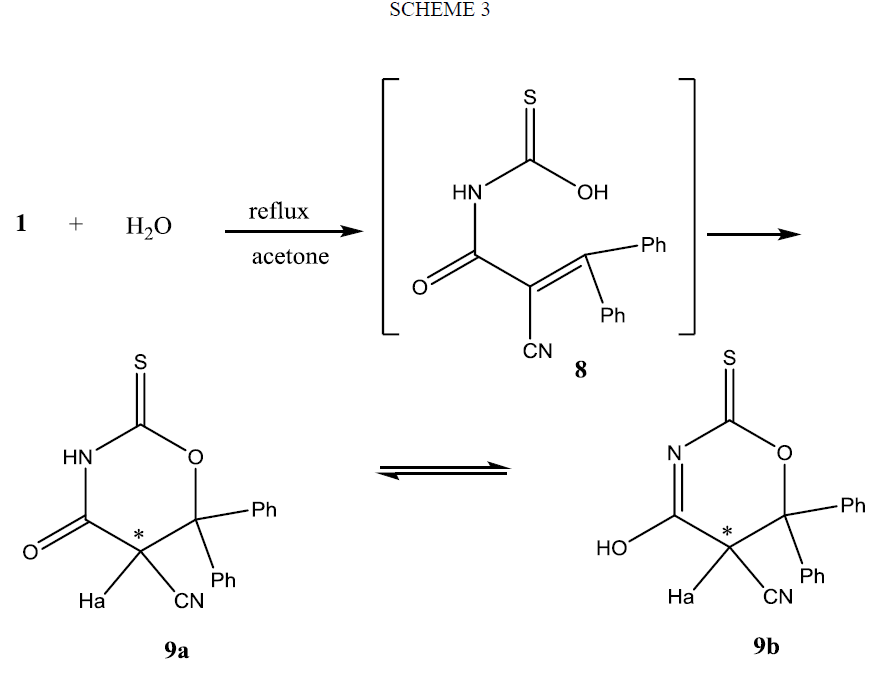
The formation of compound 9 can be rationalized on the basis of nucleophlic addition of OH group of the non-isolable adduct 8 on β-carbon atom as shown Scheme 3. Configurational assignment of 9 showed it is existed in two enantiomeric forms, since as the result of cyclization of the derivative of thiocarbamic acid 8 to oxazine derivative 9 an asymmetric carbon atom was created. 1HMNR spectrum of 9 supports the presence of methine proton Ha in two different environments. Also it shows that compound 9 exists keto-enol equilibrium 9a and 9b as it reveals two brood signals correspond to one proton, in the downfield region for NH or OH protons.
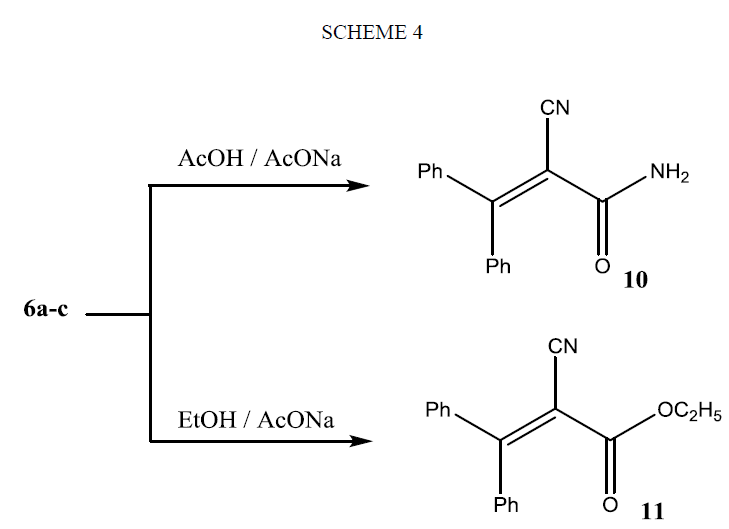
Refluxing of compounds 6(a-c) in glacial acetic acid in the presence of fused sodium acetate gave the 2-cyano-3, 3-diphenylacrylamide (10), on the other hand, they afforded an ester 11 when they refluxed in absolute ethanol in the presence of sodium acetate as shown in Scheme 4. The structures of compounds 10 and 11 were evidenced from micro analytical and spectral data (see Experimental section).
Results and Discussion
Mass spectroscopy
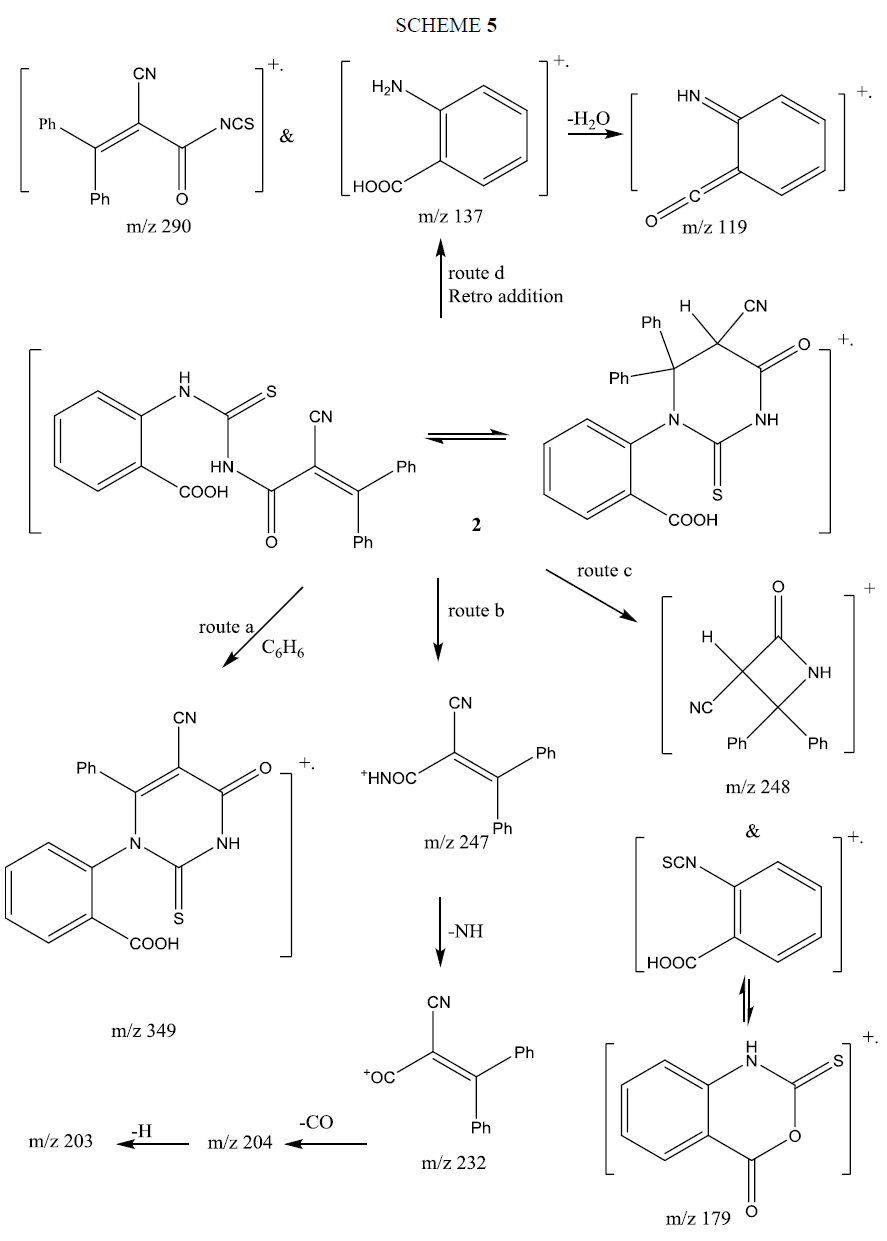
The mass spectrum of compound 2 didn't show the molecular ion peak, however, the MS peaks obtained for 2 correspond very well with its fragmentation pattern. The main fragmentation pathway of compound 2 is summarized in Scheme 5. The detection of both complementary fragments of the cleavage and rearrangement processes is attributed to their comparable ionization potentials. From the study of the mass spectrum of compound 2, it was found that the molecular ion is unstable and it had fragmented via four main routes (a-d). Route (a) involves rearrangement to pyrimidine structure, followed by loss of benzene molecule to yield fragment ion at m/z 349. Route (b) cleavage to ion m/z 247 that loss NH to give ion at m/z 232 that further loss CO followed by H to produce ions at m/z 204 and m/z 203 respectively. Route (c) cleavage to ions m/z 248 and anthranilic acid isothiocyanate at m/z 179. Route (d) Formation of isothiocyanate 1 at m/z 290 and anthranilic acid radical cation at m/z 137, which loss H2O to form keteimine at m/z 119.
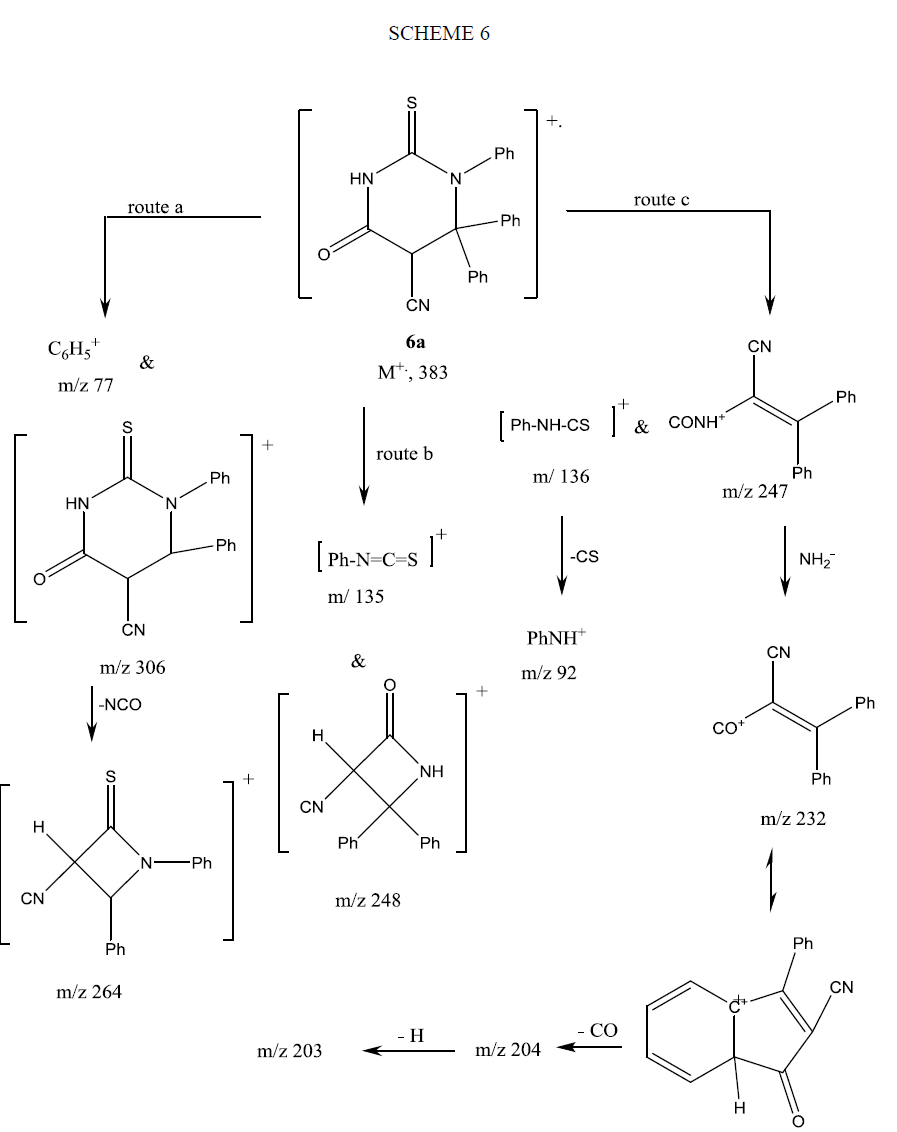
The mass spectrum of compound 6a showed its correct molecular ion peak at m/z 383 supporting its molecular formula. Its fragmentation pattern supported its structure and is given in Scheme 6. The fragmentation proceeded via three main routes. Route (a) loss of C6H5+ (m/z 77) beside fragment ion at m/z 306, the latter ion loss NCO by rearrangement of H to produce ion at m/z 264. Route (b) loss of phenyl isothiocyanate radical cation (m/z 135) and formation of ion at m/z 248. Route (c) fragmentation to PhNHCS ion at (m/z 136) and ion at m/z 247 (base peak). The latter loses NH yielding the ion m/z 232 that cyclized to indanone, which fragmented by loss of CO, followed by H producing the ions m/z 204 and m/z 203 respectively.
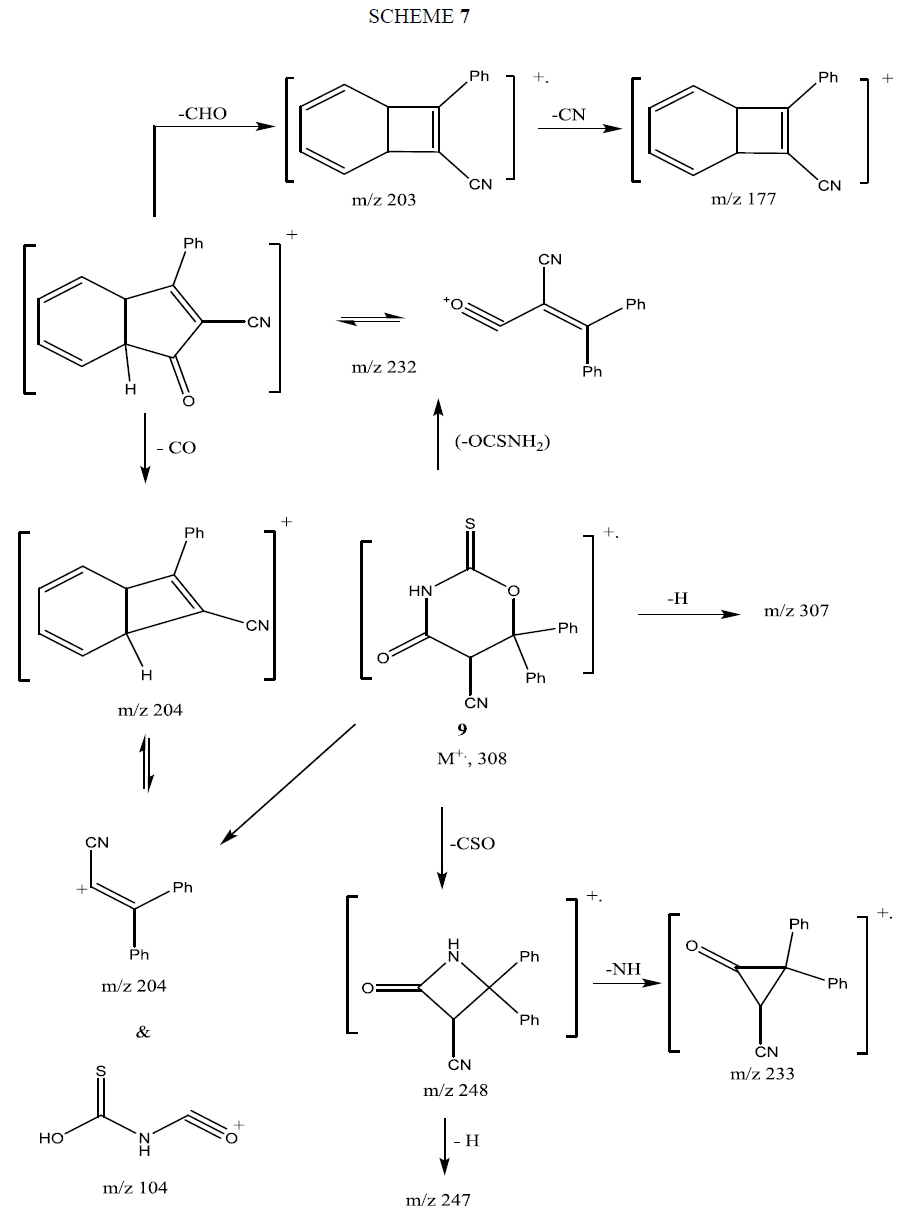
The mass spectrum of compound 9 showed its molecular ion peak at M+ 308 supporting its molecular formula. The fragmentation pattern of compound 9 is depicted in Scheme 7.
Conclusion
Synthetic utilization of α, β unsaturated acyl isothiocyanate is based on the different reactivity of both centers against nucleophiles. It is observed from the reactions mentioned above that the nucleophlic attack proceeds entirely at the isothiocyanato group rather than the α, β unsaturated system which reflects the higher reactivity of the former. The isolation of cyclic compounds reflects the extra electron deficiency at β-carbon atom that compensates via internal cyclization of the obtained adducts during the reaction process.
References
- Hemdan MM, Fahmy AF, Ali NF, et al. Synthesis of some new heterocycles derived from phenylacetyl isothiocyanate. Chinese Journal of Chemistry. 2008;26(2):388-91.
- Hemdan MM, Elshahawi MM. Synthesis of 1, 2, 4-triazoles, imidazoles, pyrimidines, quinazolines, 1, 3, 5-triazines and 1, 3-thiazines from 3-oxo-5, 6-diphenyl-2, 3-dihydropyridazine-4-carbonyl isothiocyanate. Journal of Chemical Research. 2009;2:75-7.
- Hemdan MM. Addition-cyclisation of 3-(2-thienyl) acryloyl isothiocyanate with hydrazine derivatives as a source of triazoles and thiadiazoles. Journal of Chemical Research. 2009;2009(8):489-91.
- Hemdan MM, Fahmy AF, El-Sayed AA. Synthesis and antimicrobial study of 1, 2, 4-triazole, quinazoline and benzothiazole derivatives from 1-naphthoylisothiocyanate. Journal of Chemical Research. 2010;34(4):219-21.
- Hemdan MM. Synthesis and antimicrobial activities of some heterocyclic systems from 2-furoyl isothiocyanate. Phosphorus, Sulfur and Silicon. 2010;185(3):620-7.
- Hemdan MM, Fahmy AF, Aly NF, et al. Utility of phthalimidoacyl isothiocyanate in synthesis of quinazolines, benzoxazoles, benzimidazoles, 1, 2, 4-triazoles and oxatriazepines. Phosphorus, Sulfur and Silicon and the Related Elements. 2012;187(2):181-9.
- Hemdan MM, Abou Elmagd WS, Samy SS, et al. Dodecanoyl thiosemicarbazide derivatives as useful synthons in the synthesis of 1, 2, 4-triazole, 1, 3, 4-thiadiazole and 1, 3-benzothiazole derivatives. Synthetic Communications. 2016;46(8):710-8.
- Hemdan MM, El-Mawgoude A, Heba K. Use of di-substituted thiosemicarbazide in synthesis of new derivatives of 1, 3, 4-thiadiazole, 1, 2, 4-triazole and pyrazole with their antimicrobial evaluation. Journal of Chemical Research. 2016;40(6):345-50.
- Hemdan MM, El?Sayed AA. Synthesis of Some new heterocycles derived from novel 2?(1, 3?Dioxisoindolin?2?yl) benzoyl isothiocyanate. Journal of Heterocyclic Chemistry. 2016;53(2):487-92.
- Hemdan MM, El-Bordany EA. Use of dodecanoyl isothiocyanate as building block in synthesis of target benzothiazine, quinazoline, benzothiazole and thiourea derivatives. Chemical Papers. 2016;70(8):1117-25.
- Hemdan MM, El-Mawgoude HK. Synthesis and antimicrobial evaluation of thieno [2, 3-d]-pyrimidine, Thieno [2′, 3′: 4, 5] pyrimido [1, 2-a] [1, 3, 5] triazine, Thieno [2, 3-d]-1, 3-thiazine and 1, 2, 4-Triazole Systems. Chemical and Pharmaceutical Bulletin. 2015;63(10):812-8.
- Dzurilla M, Kristian P. Reaction of α, β-unsaturated acyl isothiocyanates with alkali metal hydrosulphides. Collection of Czechoslovak Chemical Communications. 1976;41(5):1388-95.
- Dzurilla M, Kristian P, Kutschy P. Synthesis of 2-amino-6-substituted 5, 6-dihydro-4H-1, 3-thiazin-4-ones from α, β-unsaturated acyl isothiocyanates. Collection of Czechoslovak Chemical Communications. 1980;45(11):2958-64.
- Dzurilla M, Kutschy P, Kristian P. Boron trifluoride-catalyzed cyclization of N-substituted-N′-(3-substituted propenoyl)-thioureas: A new synthesis of 1, 3-thiazines. Synthesis. 1985;1985(10):933-4.
- Dzurilla M, Kutschy P, Kristian P, et al. Chem Papers. 1986;40(b):829.
- Abel-Hamid HA, Fahmy AF M, Shiba SA, et al. Egypt J Chem. 1993;63:167.